Intro
- AMP guideline에 대해 알아봅니다.
AMP guideline
AMP Guideline은 2017년 AMP(Association for Molecular Pathology)에서 발표한 NGS 변이 해석 가이드라인으로 ‘Standards and Guidelines for thr Interpretation and Reporting of Sequence Variants in Cancer’ 논문으로 발표했습니다. 논문의 내용을 살펴보며 AMP 가이드라인이 어떤 원리로 동작하고 어떤 규칙을 가지고 있는지 알아봅니다.
Abstract
AMP 가이드라인은 다양한 그룹의 연구자들이 NGS로 검출된 somatic 변이에 대해 통일된 classification, annotation, interpretation, reporting convention을 정리하고 있습니다. 전문가들의 설문조사와 합의, 문헌 검토를 바탕으로 somatic 변이를 네 단계로 분류하는 4-tiered 시스템이 만들어 졌습니다.
1
2
3
4
- tier I: variants with strong clinical significance
- tier II: variants with potential clinical significance
- tier III: variants of unknown clinical significance
- tier IV: variants deemed benign or likely benign
Cancer genomics는 급격하게 발전하는 분야이므로, 치료, 진단, 혹은 예방과 관련된 변이의 임상적 중요도는 매번 재평가되어야 합니다. 유전적 변이는 AMP 가이드라인을 준수하여 리포팅 해야합니다. 임상적 권장사항은 간결해야 하며 조직학적 소견과 임상적 소견과 함께 연관되어야 합니다.
NGS 기술 발전으로 변이 분석에 들어가는 비용과 시간은 대폭 감소했습니다. 분석 가능한 변이도 SNVs(single-nucleotide variants) 뿐만 아니라 indels, CNVs(copy number variants), fusions까지 범위가 넓어졌습니다.
Tumor DNA, RNA에서 얻은 분자생물학적 profile은 암환자의 임상적 관리에 대한 가이드가 될 수 있습니다. 진단이나 예측에 필요한 정보를 제공할 수 있고, 가능한 치료방법이나 타겟 치료를 찾을 수 있도록 도움이 될 수 있습니다.
Tumor tissue 대상 실시하는 NGS 검사에 대한 광범위한 접근을 위해서 4주 동안 NGS technical 설문 조사와 NGS reporting 설문 조사를 실시했습니다. 결과는 아래와 같습니다.
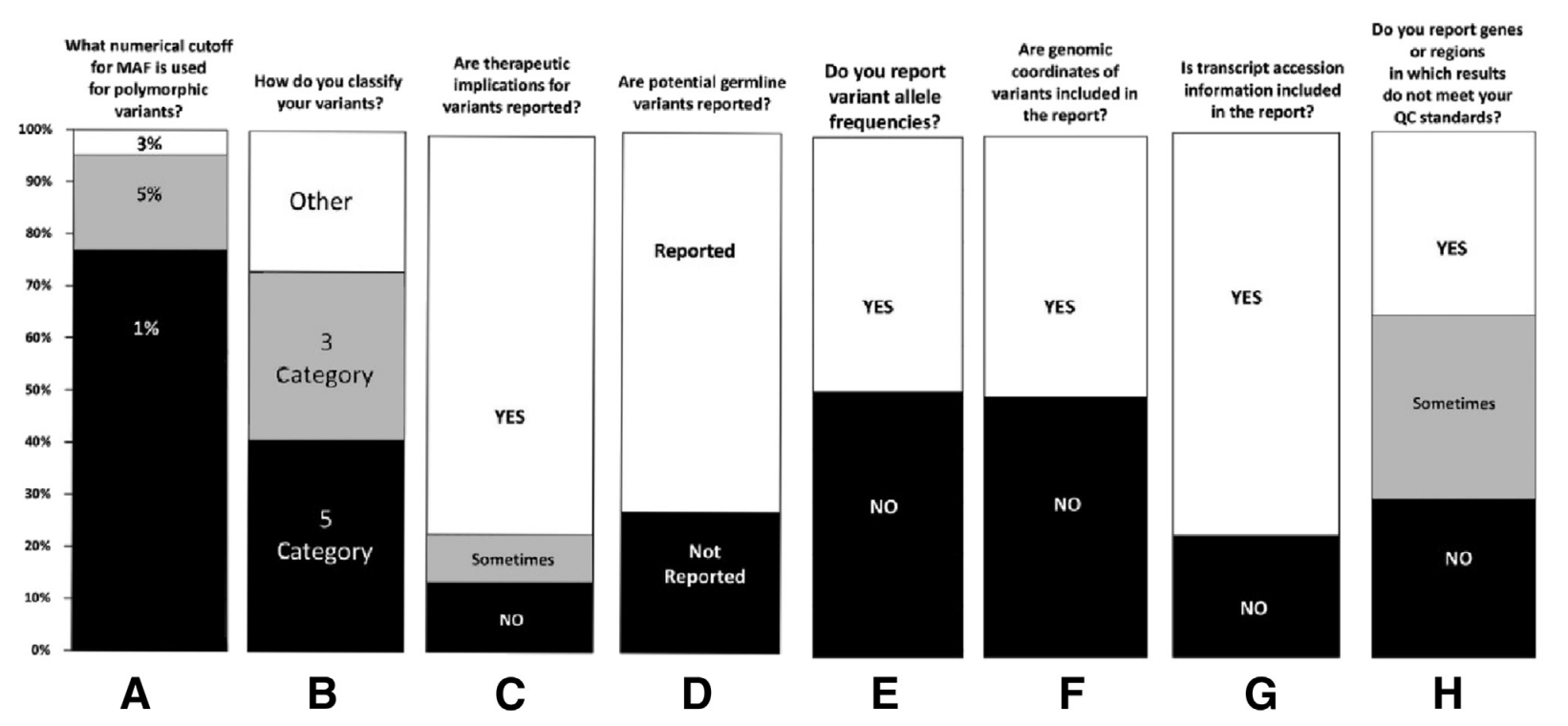 NGS technical & reporting survey
NGS technical & reporting survey
https://doi.org/10.1016%2Fj.jmoldx.2016.10.002
본 가이드라인의 목적은 cancer 대상 NGS 검사결과로 검출된 변이의 classification, annotation, interpretation, reporting의 표준방식을 수립하기 위함입니다.
Databases
Genomic Databases
다양한 tumor 종류 대상 large-scale genome sequencing project가 점점 증가하고 논문으로 발표됨에 따라 풍부한 genomic information을 바탕으로 많은 public database가 생성되고 있습니다. 예를 들어, TCGA(The Cancer Genome Atlas), COSMIC(the Catalog of Somatic Mutations in Cancer)와 같은 somatic 변이 database가 있습니다. Reference sequence information, population databases, germline variant databases 등이 분석에 자주 사용되고 있습니다. 이런 database는 somatic 변이의 정확한 annotation과 prioritization에 필수적인 정보를 제공합니다. 임상실험실은 public database 사용에 대한 아래 주의사항들을 반드시 준주해야 합니다.
1
2
3
4
5
6
1. Database content와 어떻게 만들어졌는지 이해해야 합니다. 관련 논문을 리뷰하고 database의 source, type, 목적을 이해해야 합니다.
2. Database의 한계를 이해하고 annotation 결과에 대한 과해석을 방지해야 합니다.
3. Human genome assembly 버전과 mRNA transcript reference를 확인해야 합니다. 모든 정보는 HGVS(Human Genome Variation Society) annotation 규칙을 따라야 합니다.
4. Database에 대한 불분명한 query 요청을 막기 위해서 가능하다면 HGVS nomenclature 대신 genomic coordinates를 사용합니다.
5. Database가 제공하는 genomic data의 quality를 확인합니다. the number of a specific entry, single or multiple, the depth of the study, the use of appropriate controls, confirmation of a variant's somatic origin, functional and potential drug studies 등을 확인합니다.
6. Pathological diagnosis에 대한 data quality를 확인합니다. (eg, site, diagnosis, and subtype) <br><br>
| Utility/function | Database | Location (web address) |
|---|---|---|
| Population database to </br>exclude polymorphisms | 1000 Genome Project | http://browser.1000genomes.org |
| Exome Variant Server | http://evs.gs.washington.edu/EVS | |
| dbSNP | http://www.ncbi.nlm.nih.gov/snp | |
| dbVar | http://www.ncbi.nlm.nih.gov/dbvar | |
| ExAC | http://exac.broadinstitute.org | |
| Cancer-specific </br>variant databases | Catalog of Somatic Mutations in Cancer | http://cancer.sanger.ac.uk/cosmic |
| My Cancer Genome | http://www.mycancergenome.org | |
| Personalized cancer therapy, MD Anderson Cancer Center | https://pct.mdanderson.org | |
| cBioPortal, Memorial Sloan Kettering Cancer Center | http://www.cbioportal.org | |
| Intogen | https://www.intogen.org/search | |
| ClinicalTrials.gov | https://clinicaltrials.gov | |
| IARC(WHO) TP53 mutation database | http://p53.iarc.fr | |
| Pediatri Cancer Genome Project(St. Jude Children’s Research Hospital-Washington University) | http://explorepcgp.org | |
| International Cancer Genome Consortium | https://dcc.icgc.org | |
| Sequence repositores </br>and data hosts | NCBI Genome | http://www.ncbi.nlm.nih.gov/genome |
| RefSeqGene | http://www.ncbi.nlm.nih.gov/refseq/rsg | |
| Locus Reference Genomic | http://www.lrg-sequence.org | |
| UCSC table browser | https://genome.ucsc.edu/cgi-bin/hgTables | |
| Ensemble BioMart | http://useast.ensembl.org/biomart/martview | |
| Other disease/mutation databases </br>useful in the context of variant interpretation </br>for cancer genomics | ClinVar | http://www.ncbi.nlm.nih.gov/clinvar |
| Human Gene Mutation Database | http://www.hgmd.org | |
| Leiden Open Variation Database | http://www.lovd.nl | |
| dbNSFP(compiled database of precomputed in silico prediction scores for nonsynonymous SNVs) | https://sites.google.com/site/jpopgen/dbNSFP | |
| Ensemble Variant Effect Predictor | http://www.ensembl.org/info/docs/tools/vep/index.html |
Reference Sequence Databases
Reference sequence database는 human genome assembly에 대한 version과 genomic coordinates와 같은 관련 정보를 제공합니다. 유전자에 대한 variant location mapping(coding, noncoding, untranslated region, and splice site)과 strand representation(positive versus negative)도 database로부터 계산할 수 있습니다. RefSeq, Ensembl, Locus Reference Genomic database를 사용할 수 있습니다.
Population Databases
지리학적으로 분리된 population을 대표하는 개인들의 유전자 분석결과에서 특정 locus의 alternative(minor) alleles의 빈도를 제공하는 database입니다. 주로 유전자 분석결과 polymorphic/benign으로 보이는 변이를 filter out 할 때 사용합니다. 이 때 기준점을 MAF(minor allele frequency)로 사용하는데, 수치는 1%(0.01)로 잡는 것을 권장하며 규정된 값은 아닙니다. 해당 database를 사용할 때 주의해야 할 사항 중 하나는 study 참여 당시 대상자는 모두 건강하고 질병이 없는 상태로 간주했다는 점입니다. 게다가 몇몇 전통적으로 cancer-associated로 알려진 targetable somatic 변이가 일부 population database 에서는 germline 변이로 포함되어 있습니다.
예를 들어, NM_004972.3(JAK2):c.1849G>T(c.V617F)는 myeloproliferative neoplasms에서 somatic 변이로 잘 알려져 있으며, FDA 승인 약물인 JAK(Janus kinase) inhibitor의 target으로 알려져 있습니다. 이처럼 hematological malignancies에서 검출된 변이를 평가할 때는 특별히 많은 주의를 기울어야 하는데, leukemia와 myeloodysplastic syndromes에서 발견되는 일반적인 유전자 변이가 blood 내에서 somatically mutated 되었을 가능성이 있기 때문입니다.
Cancer-Specific Databases
여러 가지 cancer 종류와 subtypes에 걸쳐 sequence variants의 발병률과 유병률에 대한 정보를 제공하는 database입니다.
Internal(Laboratory-Generated) Databases
임상검사실은 검사실 내 변이를 추적하고 일관적으로 변이 annotation 정보를 제공하기 위해 well-annotated in-house database를 구축하는 것이 중요합니다. Cancer type별로 변이의 빈도를 확인하면, sequencing alignment artifacts나 false-positive 의심 변이를 확인할 수 있습니다.
In Silico(Computational) Prediction Algorithms
In silico 예측 알고리즘은 유전자에서 발생한 nucleotide change가 단백질의 구조와 기능에 변화를 일으킬 수 있는지 예측하는데 주로 사용합니다. 분석 tool은 크게 두 가지 종류로 나뉘는데, 단백질 기능에 있어서 missense 변이가 미치는 영향력 예측, 그리고 splicing site에서 sequence variant가 미치는 영향력 예측입니다.
| Utility/function | Algorithm/software | Location (web address) |
|---|---|---|
| Missense SNV | PolyPhen2 | http://genetics.bwh.harvard.edu/pph2 |
| SIFT | http://sift.jcvi.org | |
| MutationAssessor | http://mutationassessor.org | |
| MutationTaster | http://www.mutationtaster.org | |
| PROVEAN | http://provean.jcvi.org/index.php | |
| Condel | http://bg.upf.edu/blog/2012/12/condel-for-prioritization-of-variants- involved-in-hereditary-diseases-and-transfic-for-cancer | |
| CoVEC | https://sourceforge.net/projects/covec/files | |
| CADD | http://cadd.gs.washington.edu | |
| GERP++ | http://mendel.stanford.edu/sidowlab/downloads/gerp/index.html | |
| PhyloP and PhastCons | http://compgen.bscb.cornell.edu/phast | |
| Splice site prediction | Human Splicing Finder | http://www.umd.be/HSF3 |
| MaxEntScan | http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html | |
| NetGene2 | http://www.cbs.dtu.dk/services/NetGene2 | |
| NNSplice | http://www.fruitfly.org/seq_tools/splice.html | |
| GeneSplicer | http://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml |
Variant Identification and Annotation
변이 확인은 변이 해석 과정에 있어서 중요한 시작점입니다. Variant calling 결과는 전형적인 결과 포맷 중 하나로 표현합니다; VCF(variant call format), gVCF(genomic VCF), GFF(generic feature format)
Variant annotation 또한 somatic sequence 변이를 정확하게 해석하기 위해 필요한 중요한 과정입니다. 난제 중 하나는 변이의 genomic coordinates(chromosome and position)를 이에 상응하는 cDNA/amino acid coordinate system(c. and p. syntax, respectively)로 변환하는 것입니다. 특히 alignment에 있어서 난해함이 있는 indel 변이에서 문제가 되는데, HGVS system에서는 right-aligned(더 이상 옮기지 못할 때까지 변이의 시작점을 오른쪽으로 이동시키는 것) 표현을 권장하지만 VCF는 left-aligned 표기를 필요로 합니다.
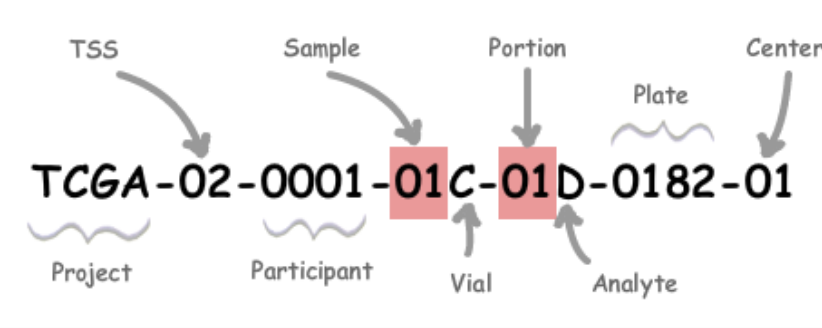 TCGA Sample Annotation
TCGA Sample Annotation
ttps://www.laidd.org/local/ubonline/view.php?id=141&group=1&returnurl=aHR0cHM6Ly93d3cubGFpZGQub3JnL2xvY2FsL3Vib25saW5lL2luZGV4LnBocD9vcmRlcnR5cGU9cmNfZCZrZXl3b3JkPUJJRyZwcm9ncmVzcyU1QiU1RD0xMyZlbnJvbF9zdGFydD0mZW5yb2xfZW5kPSZzdHVkeV9zdGFydD0mc3R1ZHlfZW5kPQ==
Standard quality metric을 제시한 논문 중 하나로, Earth BioGenome Project의 Supplementary Figure S1에 자세히 기술되어 있습니다.
x score: \(10^{x}\) = N50 contig length in kb. 만약 x가 3이라면 contig의 N50이 1,000kb임을 의미합니다.
y score: \(10^{y}\) = N50 scaffold length in kb. 만약 y가 1이라면 scaffold의 N50이 10kb임을 의미합니다.
z score: contig, scaffold 제작 과정에 문제가 없었는지 얼마나 자세히 확인했는가 나타내는 지표입니다. 3단계(0, 1, 2)로 표현합니다.
해당 논문에서 high-quality reference assembly standard를 ‘2.3.2.QV40’으로 제시하고 있습니다. 이는,
contig N50 100kb 이상
scaffold N50 1,000kb 이상
길이로 따졌을 때 90% 이상의 contig가 scaffold에 포함되고 fusion/fission/translocation 등이 다른 실험 방법으로 확인됐는지 검증한 것
을 의미합니다.
Contig Length
N50은 quality metric 중 하나로 read/contig/scaffold에 모두 적용됩니다.
우선 read/contig/scaffold를 길이가 긴 것부터 정렬합니다. 그 길이를 순차적으로 하나씩 더했을 때, 전체 길이의 절반이 넘는 순간 그에 해당하는 read/contig/scaffold의 길이를 N50으로 정의합니다. 그림에서 전체 길이는 135bp이며 50%는 약 68bp입니다. 길이가 긴 것부터 정렬한 뒤 68bp를 넘는 시점의 read/contig/scaffold는 30bp입니다. 즉, N50은 30bp입니다.
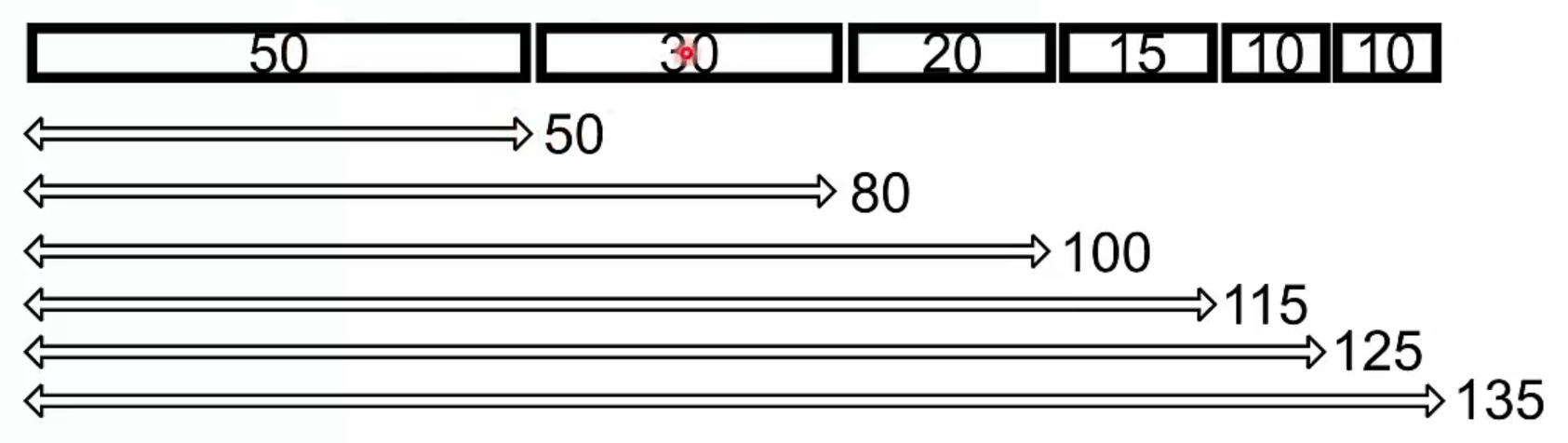 N50
N50
https://www.edwith.org/longread-seq-2023/lecture/1475113
NGx plot은 각 contig의 길이(=y축: contig나 scaffold length)와 누적 합을 전체 genome length로 나눈 비율(=x측: cumulative coverage)로 표현한 plot입니다.)
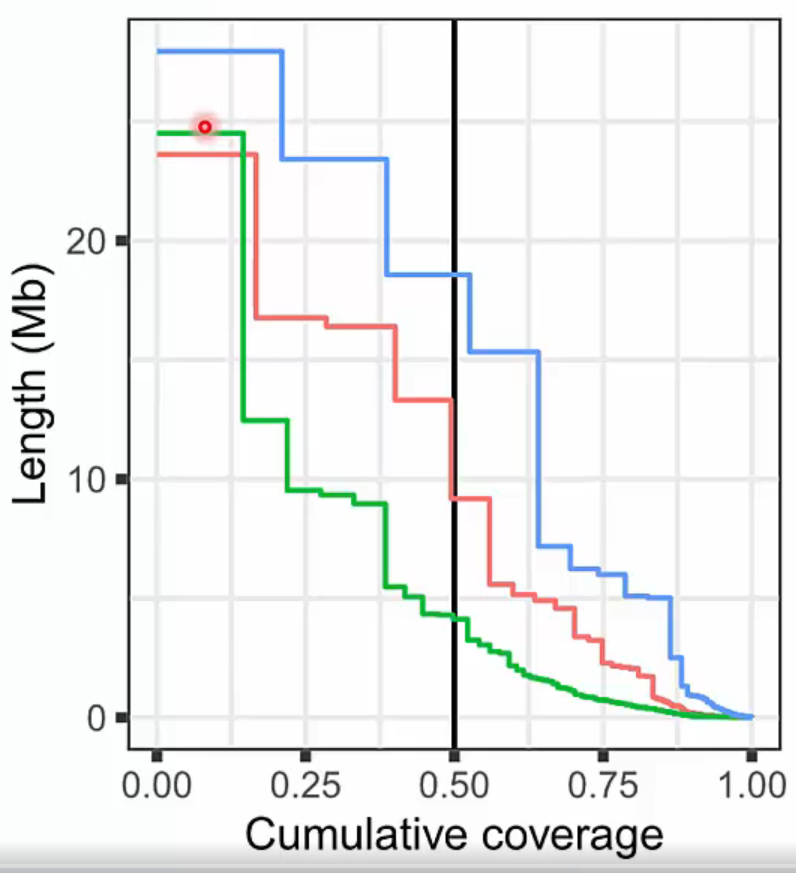 NGx Plot
NGx Plot
https://www.edwith.org/longread-seq-2023/lecture/1475113
BUSCO
BUSCO(Benchmarking Universal Single-Copy Orthologs)는 기존에 알려진 lineage-specific single-copy ortholog 유전자들이 제대로 assembly 됐는지 확인하는 tool입니다.
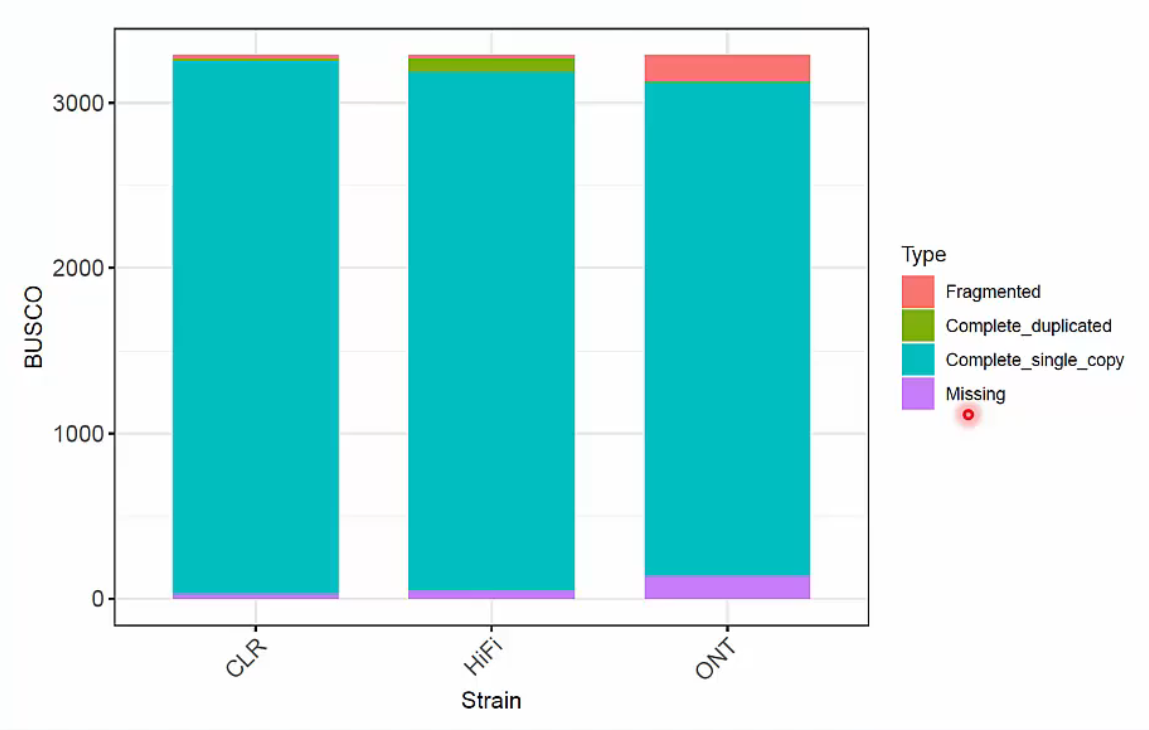 BUSCO Plot
BUSCO Plot
https://www.edwith.org/longread-seq-2023/lecture/1475113
Take Home Message
Contig length 기반으로 assembly quality를 비교할 수 있습니다.
BUSCO를 활용할 수 있습니다.