본 post는 국가생명연구자원정보센터(KOBIC) 주관 광주과학기술원(GIST) 박지환 교수님의 단일세포 분석 소개 및 실험 디자인을 정리한 내용입니다.
Intro
Single cell analysis의 필요성 및 연구배경, 실험 디자인에 대한 내용을 이해합니다.
 Single Cell Analysis 필요성
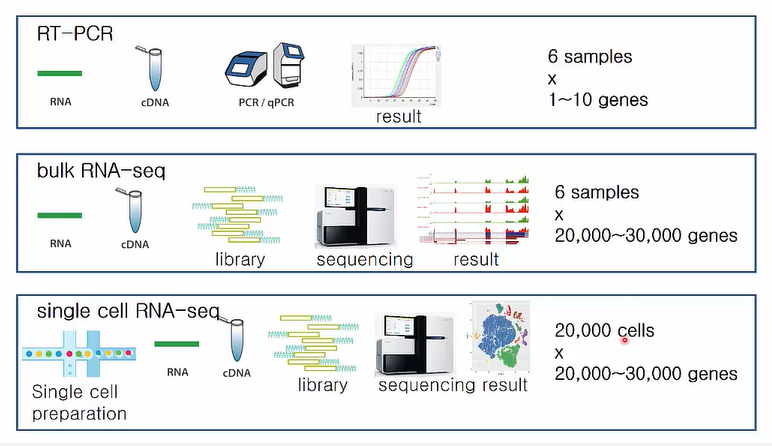
Single Cell Analysis 필요성
https://www.edwith.org/single-cell/joinLectures/361836
과거에는 RT-PCR, bulk RNA-seq 등으로 cell에서 gene expression level을 확인했습니다. 그 결과 structure, location, function, a small number of markers 등에 따라 사람은 약 200여 개 종류의 서로 다른 cell types이 존재한다고 알려졌습니다. 하지만 single cell analysis를 통해서 동일한 organ이라도 서로 다른 cell type이 존재함을 확인했습니다. 또한 한 사람당 약 20,000개의 cell과 그 곳에 포함된 약 20,000~30,000개의 gene에 대해 분석하고 더 정확한 결과를 얻을 수 있게 되었습니다.
Single Cell Analysis 필요성
 Single Cell Analysis 필요성
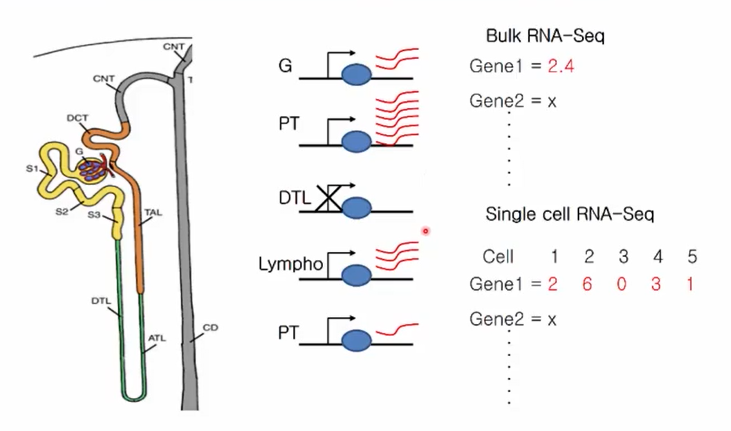
Single Cell Analysis 필요성
https://www.edwith.org/single-cell/joinLectures/361836
RNA-seq은 cell을 구분하지 못하고 평균적인 gene expression level을 확인합니다.
반면 single cell analysis는 cell별로 gene exporession level을 구분하여 확인할 수 있습니다.
 Single Cell Analysis 필요성
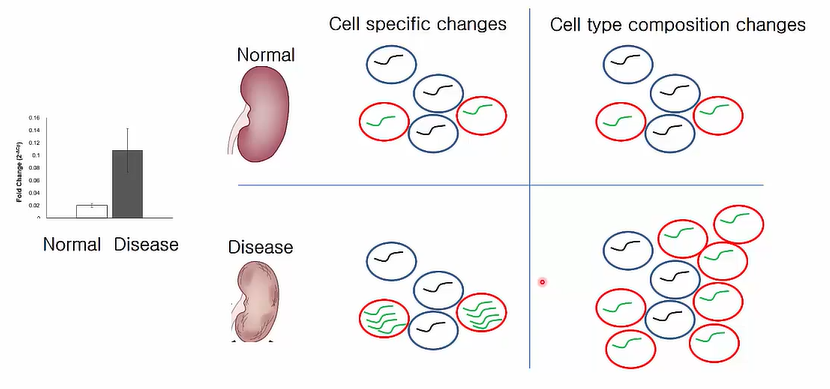
Single Cell Analysis 필요성
https://www.edwith.org/single-cell/joinLectures/361836
또한 single cell analysis를 활용하면 cell specific change와 cell type composition change를 구분할 수 있습니다. 예를 들어 정상인과 환자의 single cell analysis를 진행하면 disease와 연관된 red cell의 green gene expression high level이 cell 내 exporession 증가에 의해서인지(cell specific change), 혹은 red cell의 증가에 의해서인지(cell type coposition change) 구분할 수 있습니다.
 Single Cell Analysis 필요성
Single Cell Analysis 필요성
https://www.edwith.org/single-cell/joinLectures/361836
뿐만 아니라 cell이 분화하면서 변화하는 gene expression level을 추적 관찰할 수도 있습니다. 더 나아가 생물의 진화 양상을 추적할 수도 있습니다.
Basic Steps of Single Cell Analysis
 Single Cell Analysis 기본 단계
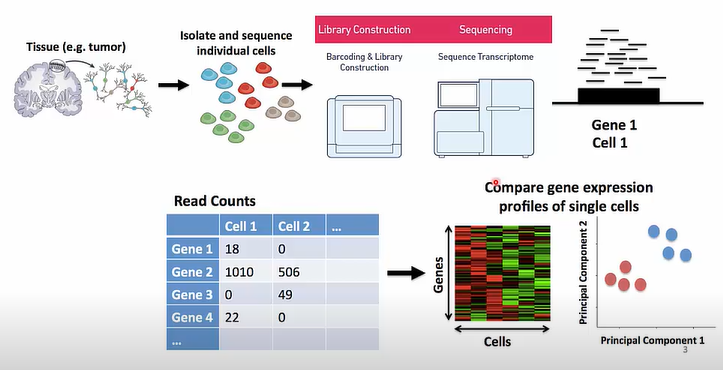
Single Cell Analysis 기본 단계
https://www.edwith.org/single-cell/joinLectures/361836
Single cell analysis의 기본적인 단계입니다. Tissue에서 cell을 분리한 다음 library를 만들고 rna seq을 진행합니다. Gene expression data를 clustering한 뒤 발현양을 비교합니다.
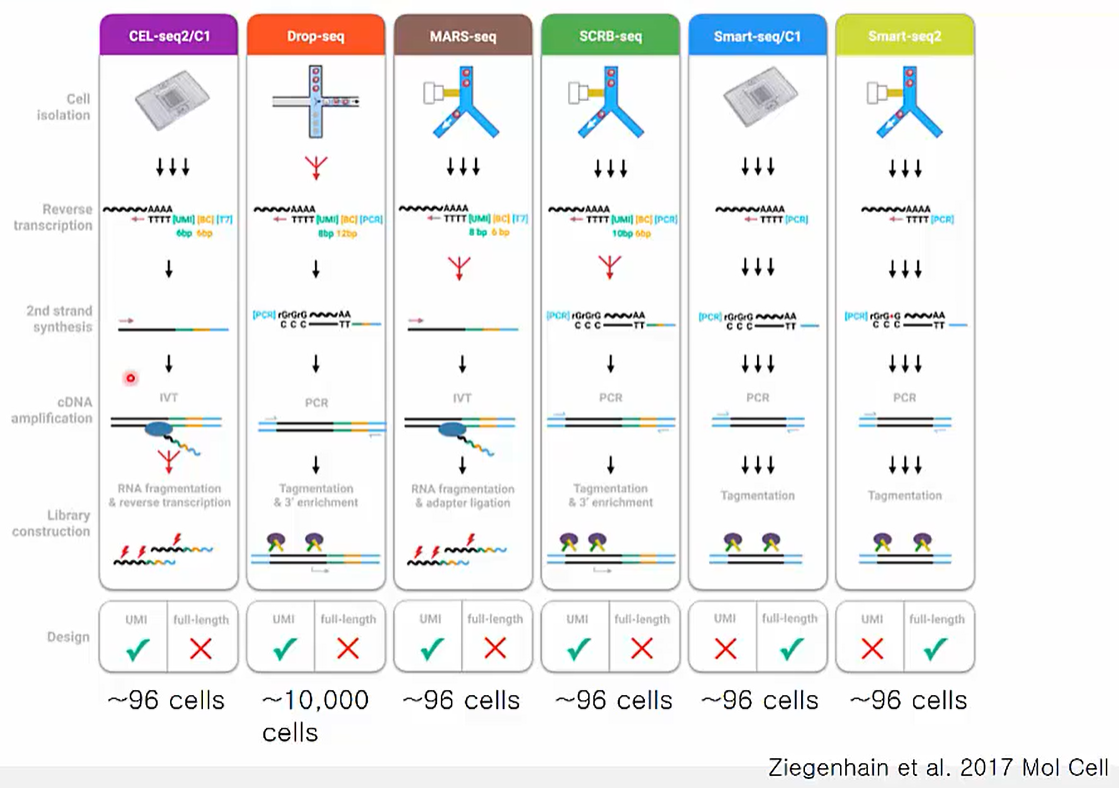 Single Cell Analysis: Library Preparation
Single Cell Analysis: Library Preparation
https://www.edwith.org/single-cell/joinLectures/361836
Library prep에 사용할 수 있는 방법은 여러 가지 개발되어 있으며 상요화 된 기술도 존재합니다. UMI 사용여부, cDNA를 얼만큼 cover하는지, 그리고 한 번에 몇 개의 cell을 cover할 수 있는지 등에 차이가 있습니다.
Experimental Design for Single Cell Analysis
기본적으로 모든 tissue에 대해 single cell analysis를 진행할 수 있습니다. 하지만 tissue 종류에 따라 dissociation 난이도가 다르고 포함하고 있는 total RNA 양도 다르기 때문에, 특성을 고려한 실험 디자인이 필요합니다.
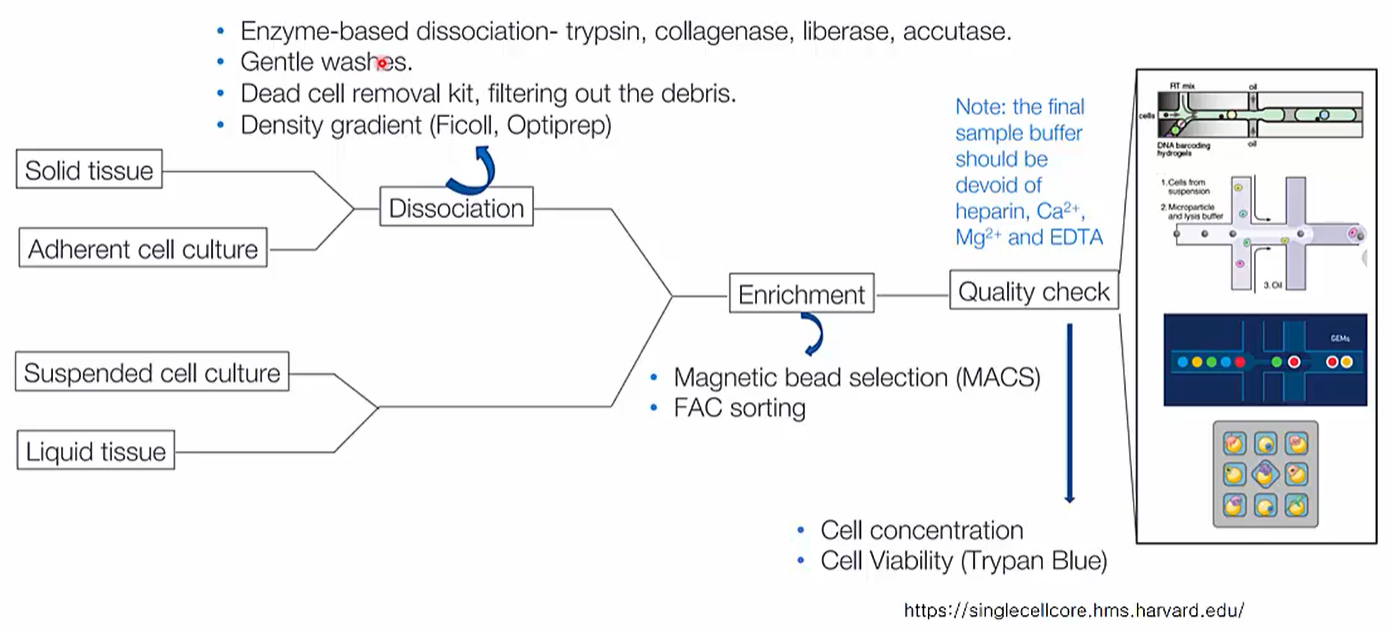 Sample Preparation Protocol Varies by Cell-types
Sample Preparation Protocol Varies by Cell-types
https://www.edwith.org/single-cell/joinLectures/361836
RNA 양이 적은 solid tissue나 adherent cell culture는 dissociation을 수행합니다. 관심있는 cell만 골라낼 필요가 있는 경우에는 MACS나 FAC sorting으로 enrichment를 수행합니다.
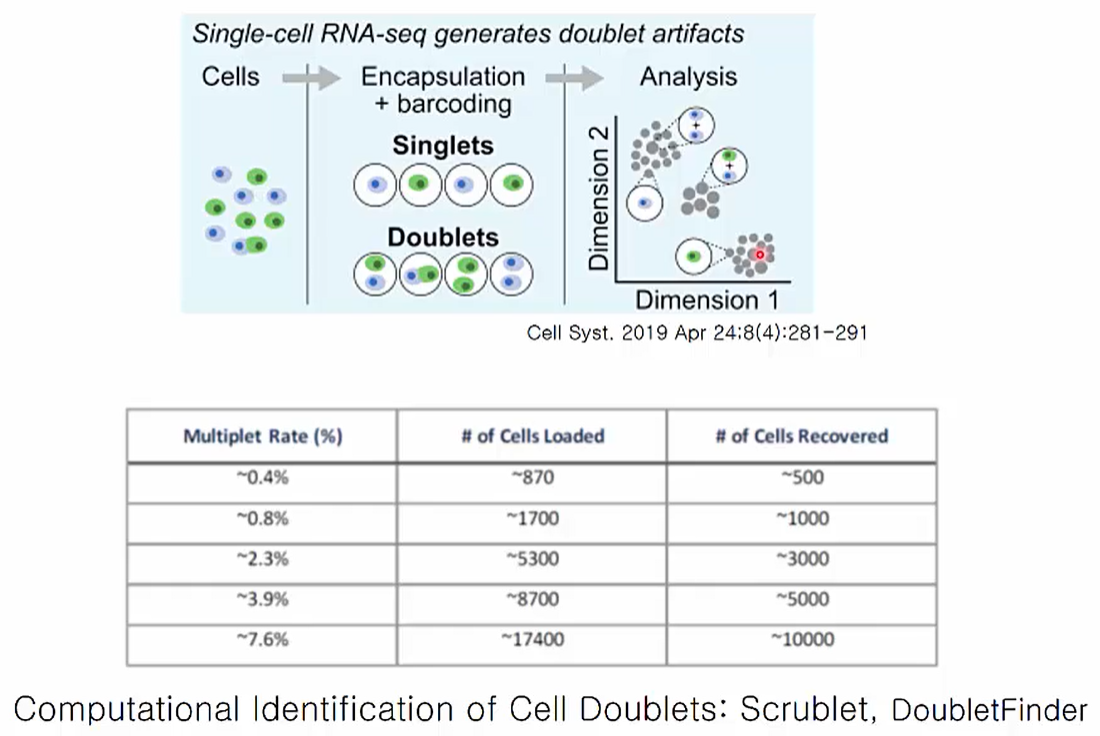 Doublets Problems
Doublets Problems
https://www.edwith.org/single-cell/joinLectures/361836
Single cell analysis의 문제점 중 하나는 바로 doublets입니다. Droplet 방식을 사용하는 single cell anlaysis는 필연적으로 doublelets 문제를 나타내는 것으로 알려져 있는데, cell loading 개수에 따라 그 비율이 증가한다고 알려져 있습니다.
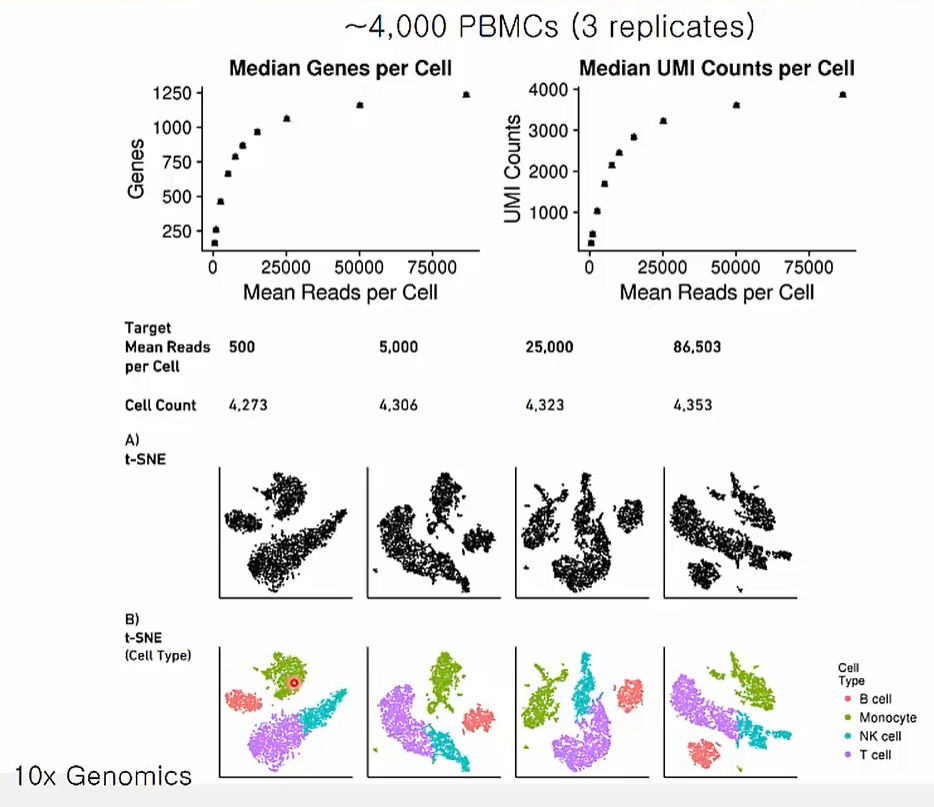 Sequencing Depth for scRNAseq
Sequencing Depth for scRNAseq
https://www.edwith.org/single-cell/joinLectures/361836
Single call analysis에 적당한 sequencing depth를 찾기 위한 data입니다. 약 30,000~50,000x에서 적당한 genes 개수와 umi 수를 확인할 수 있습니다. 물론 적당한 depth 또한 tissue types에 따라 달라질 수 있음을 고려해야 합니다.
또한 흥미로운 사실은 86,503x data에서 확인된 네 개의 cell clustering 결과가 25,000x, 5,000x, 500x로 줄였을 때 변함없이 네 개의 cluster가 나타남을 봤을 때 depth가 cell clustering에는 별다른 영향을 주지 않음을 확인할 수 있습니다.
Chanllenges and Future Direction for scRNAseq
The sparsity of data
Transcript가 적으므로 data signal보다는 noise가 더 많이 나타나는 문제가 있습니다. 따라서 발현량이 0인 gene을 다수 나타납니다. 이는 RNA capture 효율과 cDNA conversion의 문제로 나타난 결과입니다.
Batch effect
실험자, 어떤 연구실에서 실험했는지, 혹은 어떤 실험조건으로 진행했는지 등에 따라서 batch effect가 발생할 수 있습니다. 이는 computational method나 서로 다른 샘플을 multiplexing하여 해결할 수 있습니다.
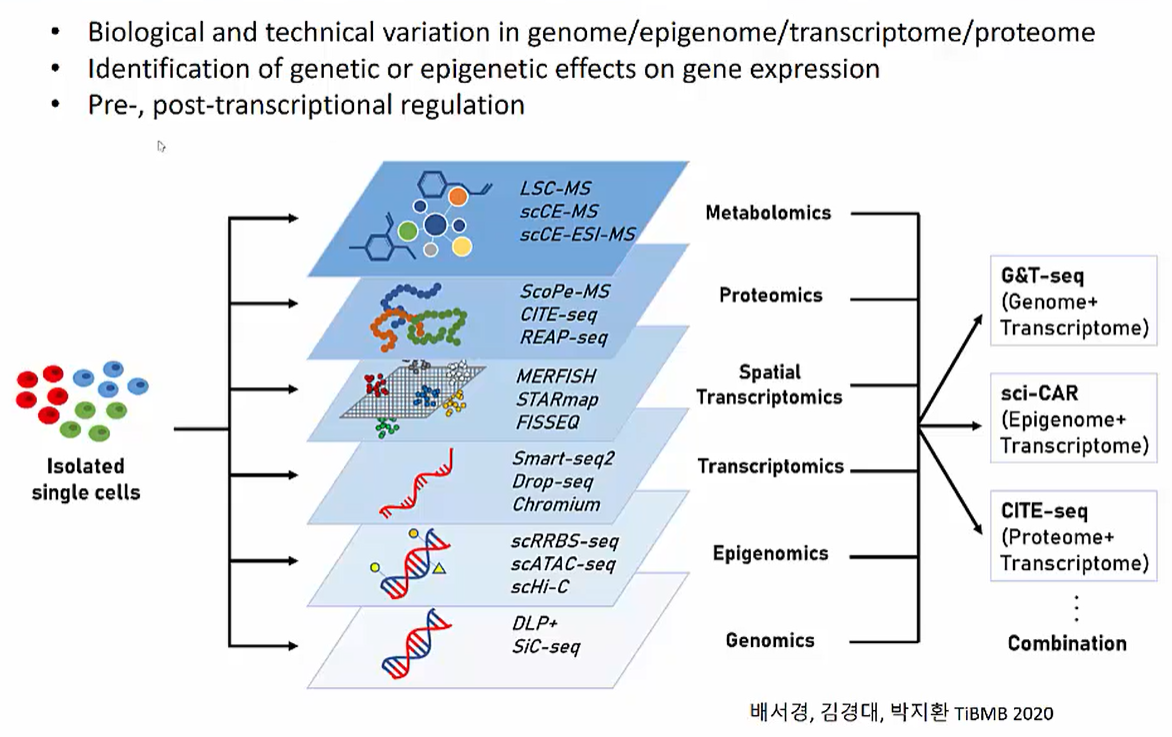 Integration of Single Cell Multi-omics Data
Integration of Single Cell Multi-omics Data
https://www.edwith.org/single-cell/joinLectures/361836
미래에는 single cell isolation 이후 genomics부터 metabolomics까지 다양한 multi-omics data를 분석하고 활용할 수 있을 것입니다.
Take Home Message
Single cell analysis의 원리와 활용 가능성에 대해 알아볼 수 있었습니다. 현재 업무에서 사용중인 genomics data와 disease, treatment와 연관된 database들이 scRNAseq 결과와 결합한다면 personalized medicine에 활용될 수 있음을 엿볼 수 있었습니다.