본 post는 국가생명연구자원정보센터(KOBIC) 주관 이화여자대학교 생명과학과 이상혁 교수님의 전사체 데이터 분석를 정리한 내용입니다.
Intro
RNA-seq data의 differential exporession, visual exploration에 대해 알아봅니다.
Differential Expression
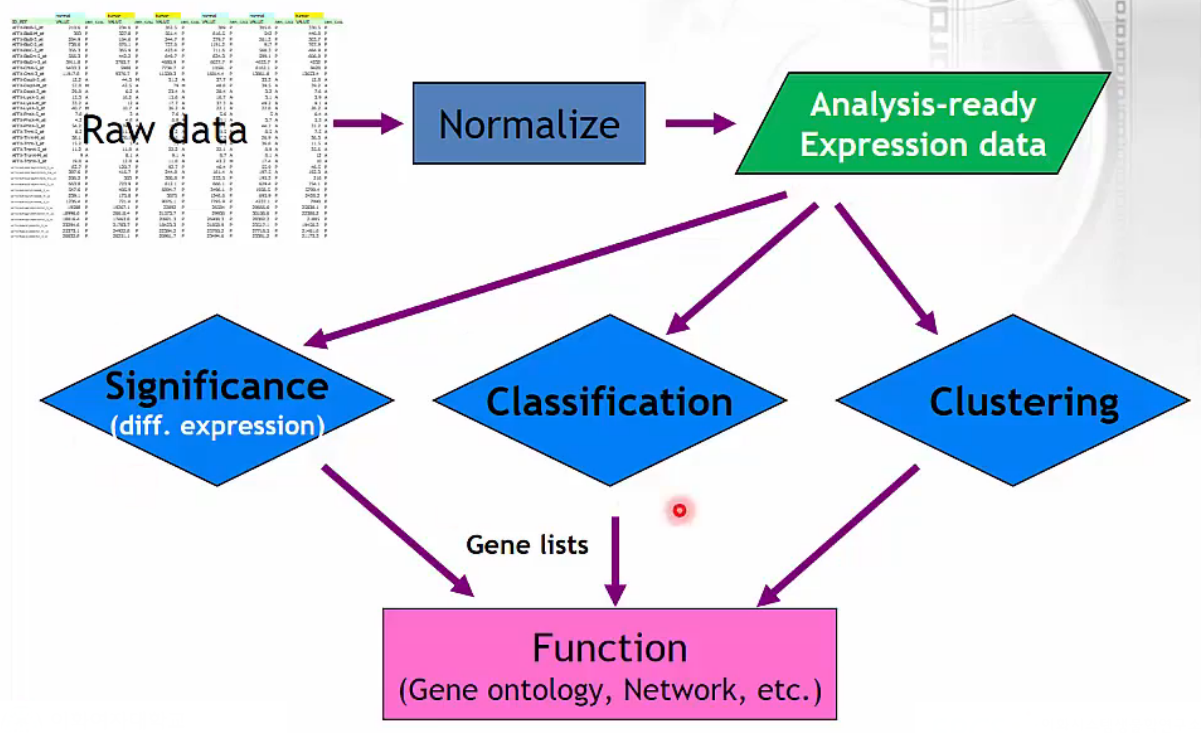 Typical Process of Transcriptome Analysis
Typical Process of Transcriptome Analysis
https://www.edwith.org/transcriptome/lecture/1382678
- 전형적인 RNA-Seq analysis 과정은 다음과 같습니다. 지난 시간 data preprocessing 및 normalization, quantification까지 알아봤습니다. 이번 차시에서는 differential expression을 알아봅니다. 다음 차시에 순차적으로 classification, clustering에 대해 알아봅니다.
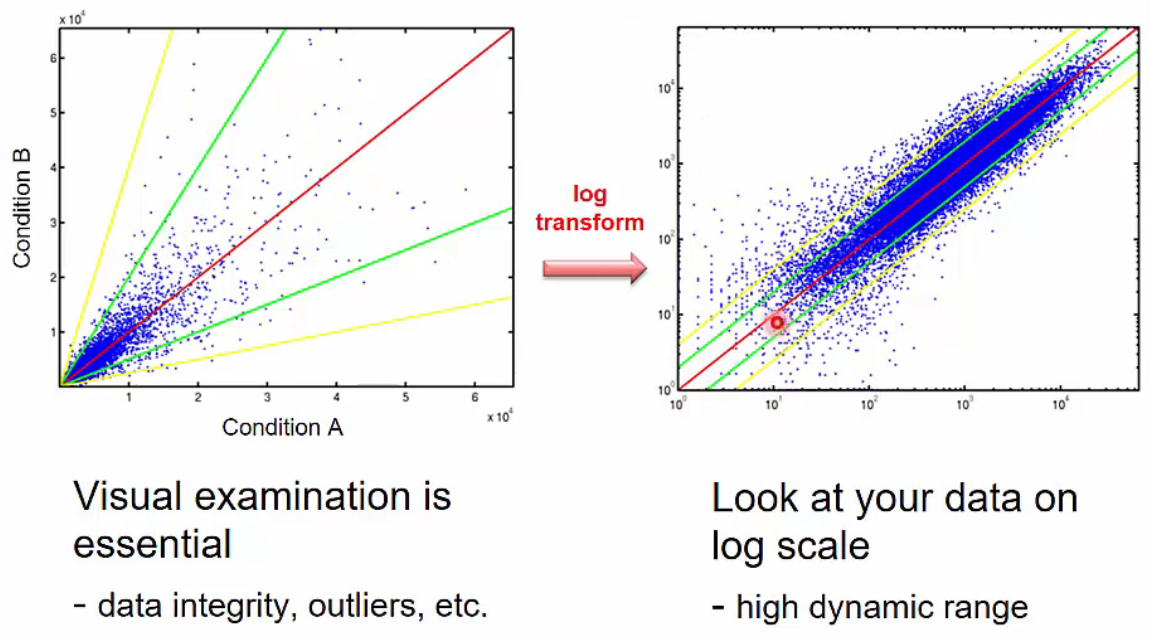 Visual Comparison of Two data sets - Scatter plot
Visual Comparison of Two data sets - Scatter plot
https://www.edwith.org/transcriptome/lecture/1382678
- 두 개의 data sets를 비교하는데 visualization은 중요합니다. Scatter plot을 그려보면 data integrity, outlieres 등을 파악하는데 도움이 됩니다.
- Gene expression range는 gene별로 큰 차이가 나기때문에(매우 작은 양부터 매우 큰 양까지) log scale에서 비교하면 보기가 좋습니다.
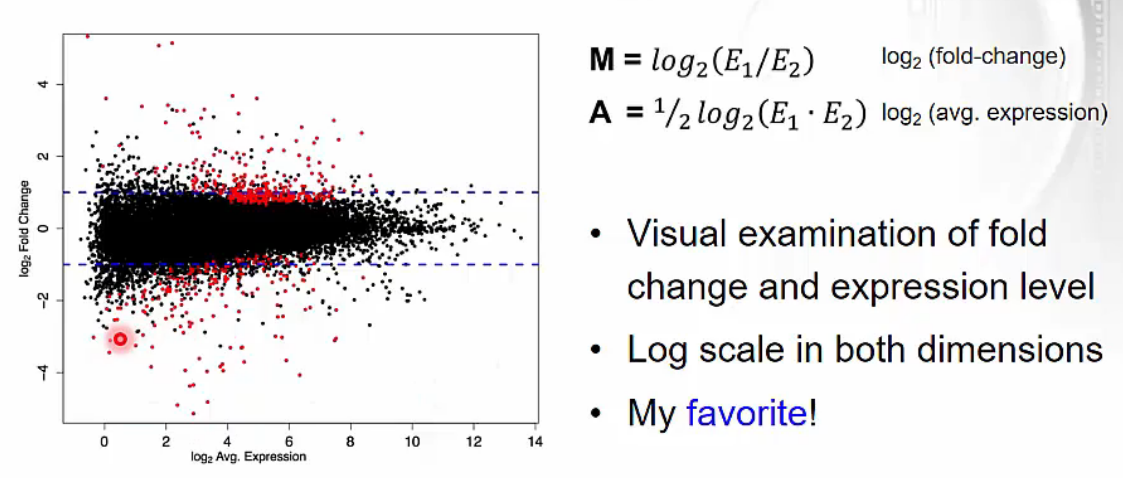 Visual Comparison of Two data sets - MA plot
Visual Comparison of Two data sets - MA plot
https://www.edwith.org/transcriptome/lecture/1382678
- MA plot으로도 두 개의 data set을 비교해서 볼 수 있습니다.
- x axis는 average express 값을 의미합니다. 즉, 오른쪽으로 갈수록 많이 expression 된다고 해석할 수 있습니다.
- y axis는 fold-change 값을 의미합니다. 즉, 위로 올라갈수록 E1이 E2에 비해 많이 expression 된다고 해석할 수 있습니다.
Differential Expression
- Differentially Expressed Genes(DEGs)는 다양한 조건에서 gene expression이 증가 혹은 감소하는 양상을 확인하는 방법입니다.
- normal vs. tumor
- treated vs. untreated
- time series profiles
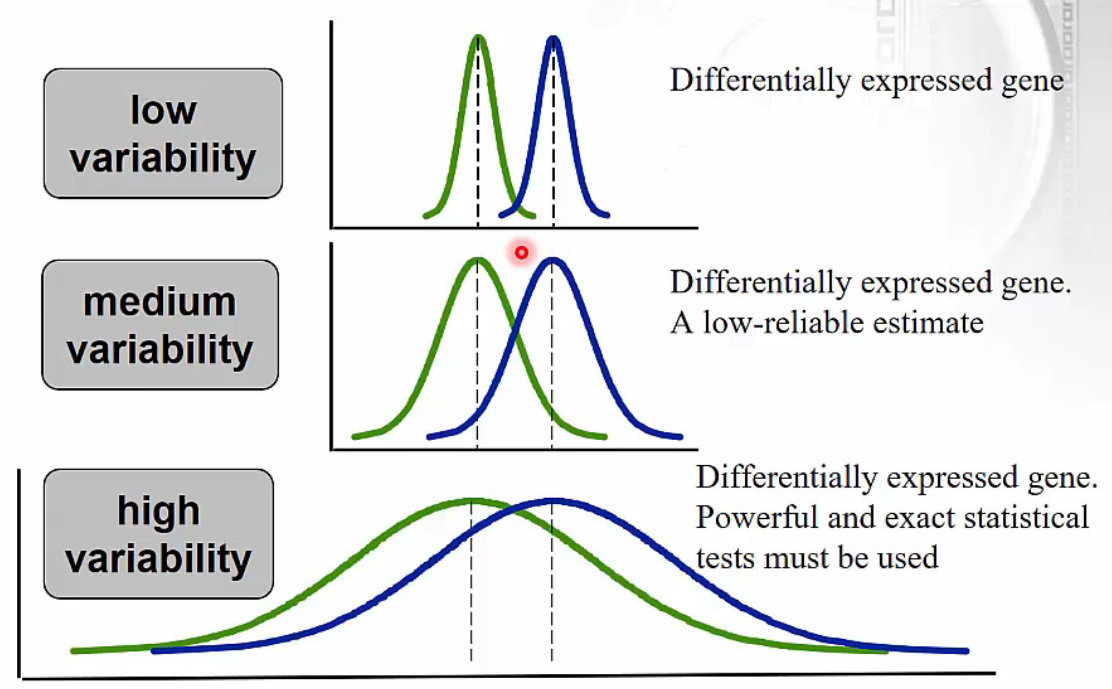 Fold change does not take variation into account
Fold change does not take variation into account
https://www.edwith.org/transcriptome/lecture/1382678
- 그림에서 두 개 genes에 대해 평균은 각각 같지만 expression 분포가 세 가지 유형으로 나뉘는 것을 확인할 수 있습니다.
- low variability: variable이 작고 두 개 genes 사이 express에 명확한 차이가 있음을 확인할 수 있습니다.
- high variability: variable이 크고 두 개 genes 사이 express에 거의 차이가 없음을 확인할 수 있습니다.
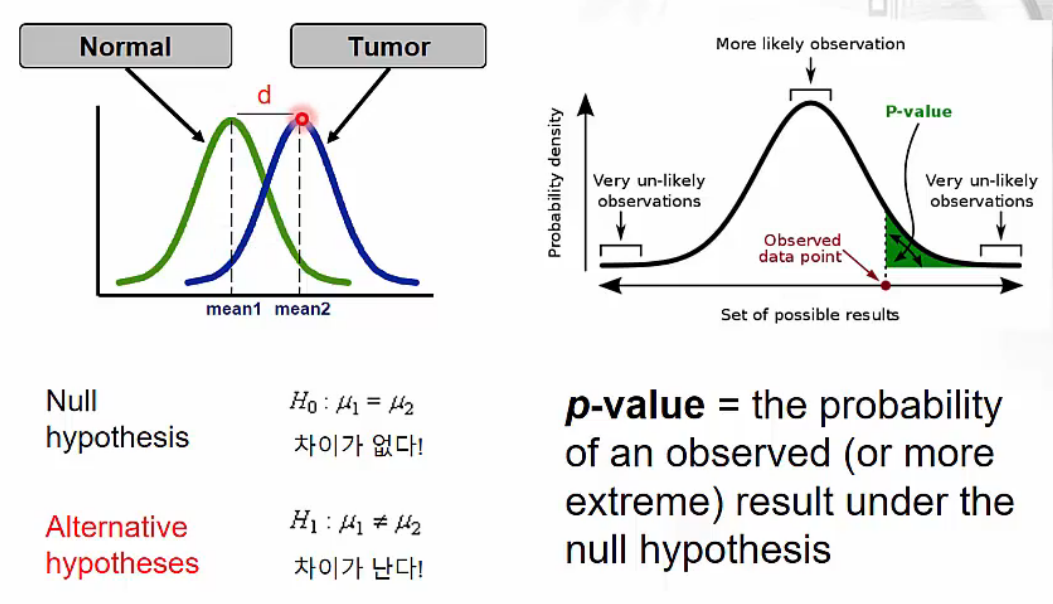 Hypothesis Testing and p-value
Hypothesis Testing and p-value
https://www.edwith.org/transcriptome/lecture/1382678
- Normal과 tumor 사이 mean 값의 차이가 있는지 없는지 확인하기 위해 hypothesis testing을 진행합니다. 두 mead 값의 차이가 없다는 null hypothesis를 세우고 p-value(오른쪽 그래프의 녹색 면적)를 계산합니다. p-value가 기준(normally 0.05)보다 낮을 때 우연한 결과가 아니라고 추정할 수 있고 null hypothesis를 기각하여 두 mean 값에 차이가 있다고 결론내릴 수 있습니다.
 t-test
t-test
https://www.edwith.org/transcriptome/lecture/1382678
- t-test는 두 개 그룹의 mean 값에 차이가 없다는 hypothesis를 test하는 방식입니다. $\frac{signal}{noise}$ 두 개 그룹 mean 값의 차이(signal)를 두 개 그룹 분포의 크기의 차이(noise, standard error)로 나눠준 값으로 계산합니다.
- 그룹이 세 개 이상일 때 t-test는 ANOVA로 변경됩니다.
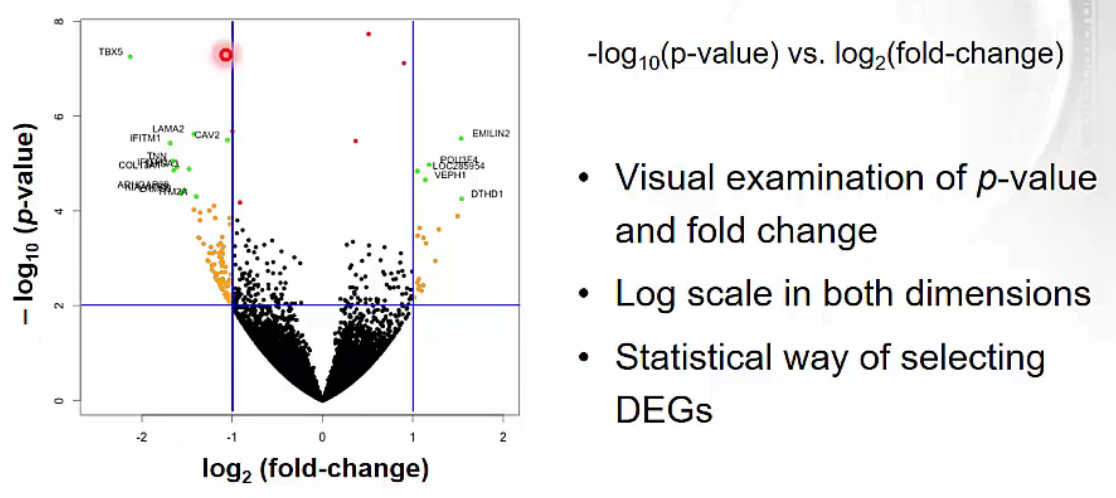 Volcano plot
Volcano plot
https://www.edwith.org/transcriptome/lecture/1382678
- p-value와 fold-change를 동시에 표현한 plot입니다.
- x축의 fold-change는 -1 미만과 1 이상이 두 배 이상 차이나는 gene으로 볼 수 있습니다.
y축의 p-value는 2 이상일 때 $10^{-2} = 0.01$이하, 곧 유의미하게 차이나는 gene으로 볼 수 있습니다.
- Multiple comparison test problem
- 생물학에서 비교반복실험은 흔히 있습니다.
- p-value 0.05 이하라는 의미는, 두 개 그룹간 차이가 존재할 확률이 95%라는 의미인 동시에 우연히 차이나는 것처럼 보일 확률이 5%라는 의미입니다.
- 예를 들어 10,000개의 gene 대상으로 p-value 5% 기준으로 잡으면 500개 gene이 우연하게 선정될 수 있습니다. (잘못된 결과)
- 10,000번의 t-test p-value 5% 기준으로 1,000개의 gene이 선택됐을 때, 결국 500/1,000 선택된 gene의 50%가 잘못된 결과로 볼 수 있습니다. (very high false discovery rate)
- 여러 번 비교반복실험을 진행할 때 단일 테스트의 p-value 5% 기준을 그대로 사용하면 이러한 문제가 발생할 수 있으므로, 따라서 multiple comparison을 위한 p-value의 보정이 필요합니다.
- Boneferroni correction
- p-value를 p-value/test counts로 보정합니다.
- 예를 들어, 1,000번의 test를 진행하면 p-value 0.05는 0.05/1,000 = 0.00005로 기준으 삼는 것입니다.
- 그러나 이 방법은 too conservative하여 많이 사용하지는 않습니다.
- False discovery rate(FDR)
- $\frac{#false positives}{#called significant}$
- Benjamini-Hochberg procedure
- 반복실험의 p-value들을 오름차순으로 정렬합니다.
- Bonferroni correction의 corrected p-value(p-value/test counts)를 2배, 3개, 4배, … , k배로 늘려가면서 기준을 만족하는 k값을 찾습니다.
- FDR approach를 통해 찾은 adjusted p-value를 q-value라고 합니다.
- 즉, q-value 5%는 significant tests 중 false positive로 확인된 비율이 5%임을 의미합니다.
- Statistical testing for differential expression
- 생물학 실험할 때 반드시 세 번 이상 반복실험 하는 것이 좋습니다.
- Normalization은 샘플간 expression 비교할 때 필수입니다.
- 분포 모델은 negative binomial distribution 사용하는 것이 바람직 합니다.
- multiple testion problems를 해결하기 위한 correction 과정은 필수입니다.
Sample Validation
- 다양한 이유로 sample이 서로 뒤바뀌는 경우가 발생할 수 있습니다. 이를 확인할 수 있는 다양한 방법이 있습니다.
- Paired sample shcek
- SNP concordance에 기반한 paired sample check 방법입니다.
- tool: NGSCheckMate, BAMixChecker
- Gender check
- X/Y chromosome 상에서 read depth나 allele frequency에 기반한 gender check 방법입니다.
- tool: SEXCMD
- Ethnicity inference
- 인종별로 나타나는 allele에 기반하여 ethnicity를 추정하는 방법으로 WGS or WES data를 대상으로 사용 가능합니다.
- tool: SeqSQC, EthSEQ, LASER 2.0
Mapping
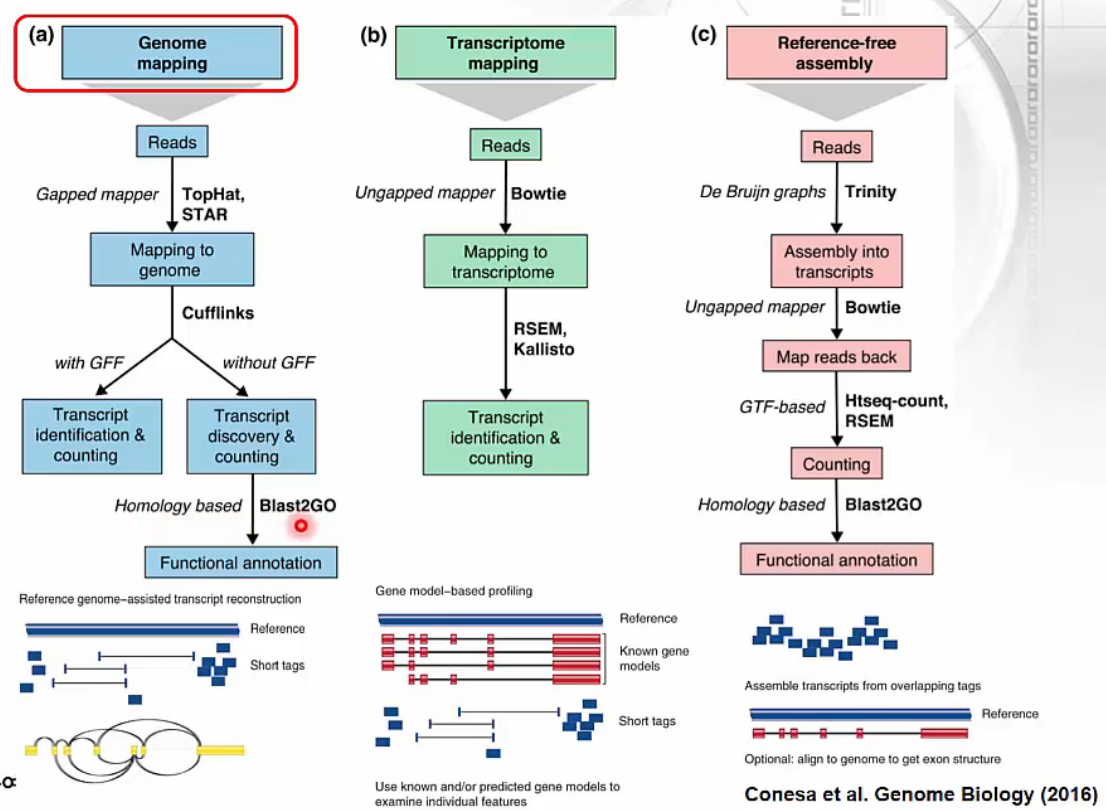 RNA-Seq Mapping
RNA-Seq Mapping
https://www.edwith.org/transcriptome/lecture/1382678
- 다양한 mapping 방법이 있습니다. 최근에는 genome에 mapping하는 방법이 주로 사용됩니다.
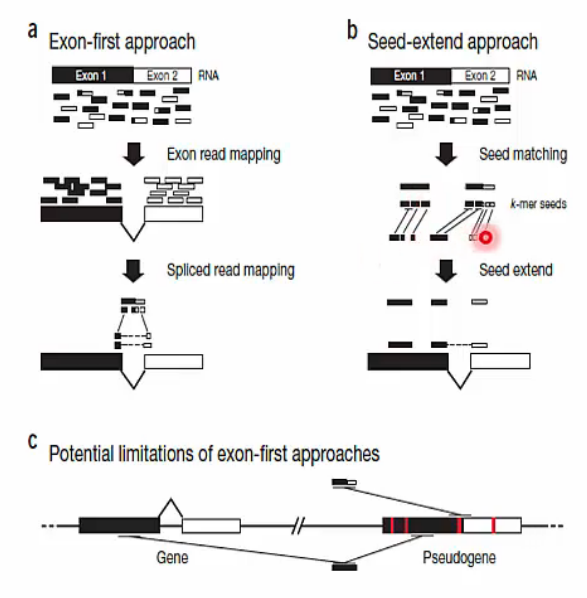 Strategies for gapped alignments
Strategies for gapped alignments
https://www.edwith.org/transcriptome/lecture/1382678
- RNA-Seq data를 genome에 mapping하는 방식은 크게 두 가지로 나눠 생각할 수 있습니다.
- Exon-first approach
- 이미 알려진 exon sequence에 read를 mapping하는 방식입니다.
- tool: MapSplice, SpliceMap, TopHat
- 하지만 gene과 pseudogene을 구분하지 못하고 모두 mapping 한다는 단점이 있습니다.
- Seed-extend methods
- N-mer의 seed reads가 어디에 matching 되는지 확인 후 확장시켜 나가는 방식입니다.
- tool: GSNAP, QPALMA
- Exon-first approach
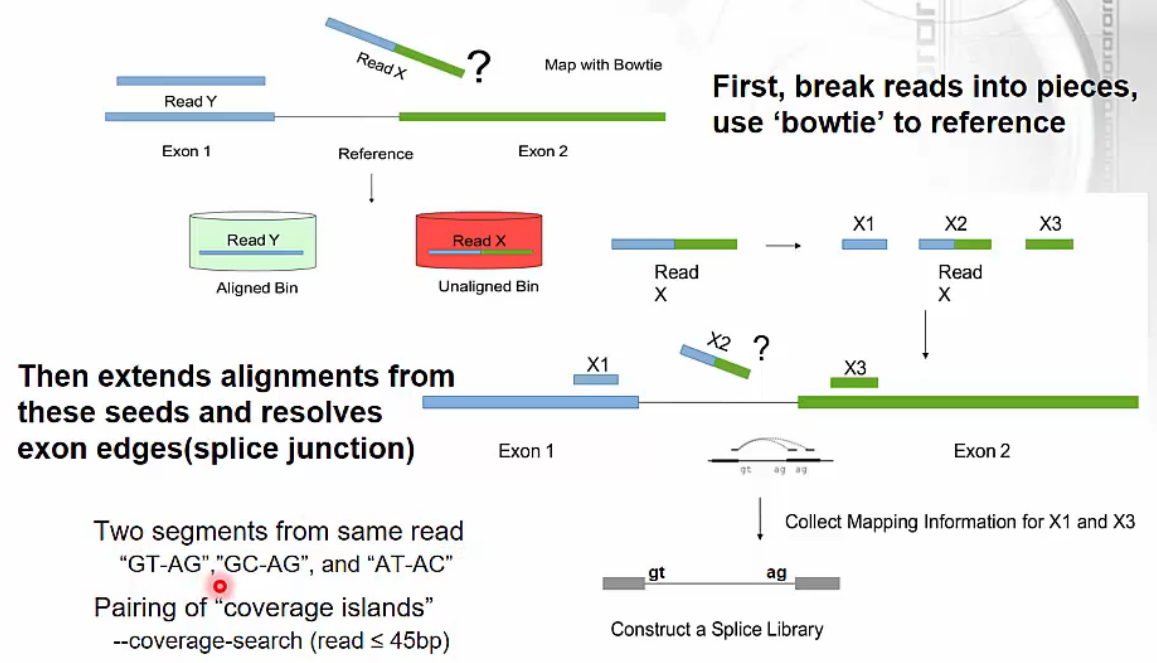 Tophat: Spliced Read Mapper
Tophat: Spliced Read Mapper
https://www.edwith.org/transcriptome/lecture/1382678
- Tophat - spliced read mapper
- Exon 내 mapping되는 reads를 먼저 선별합니다.
- 두 개의 exon에 걸쳐있는 reads는 각각 exon 영역에 맞는 영역으로 나눕니다. 이 때 canonical intron이 지니고 있는 특징적인 서열(intron의 시작과 끝 서열: GT-AG, GC-AG, AT-AC)을 활용합니다.
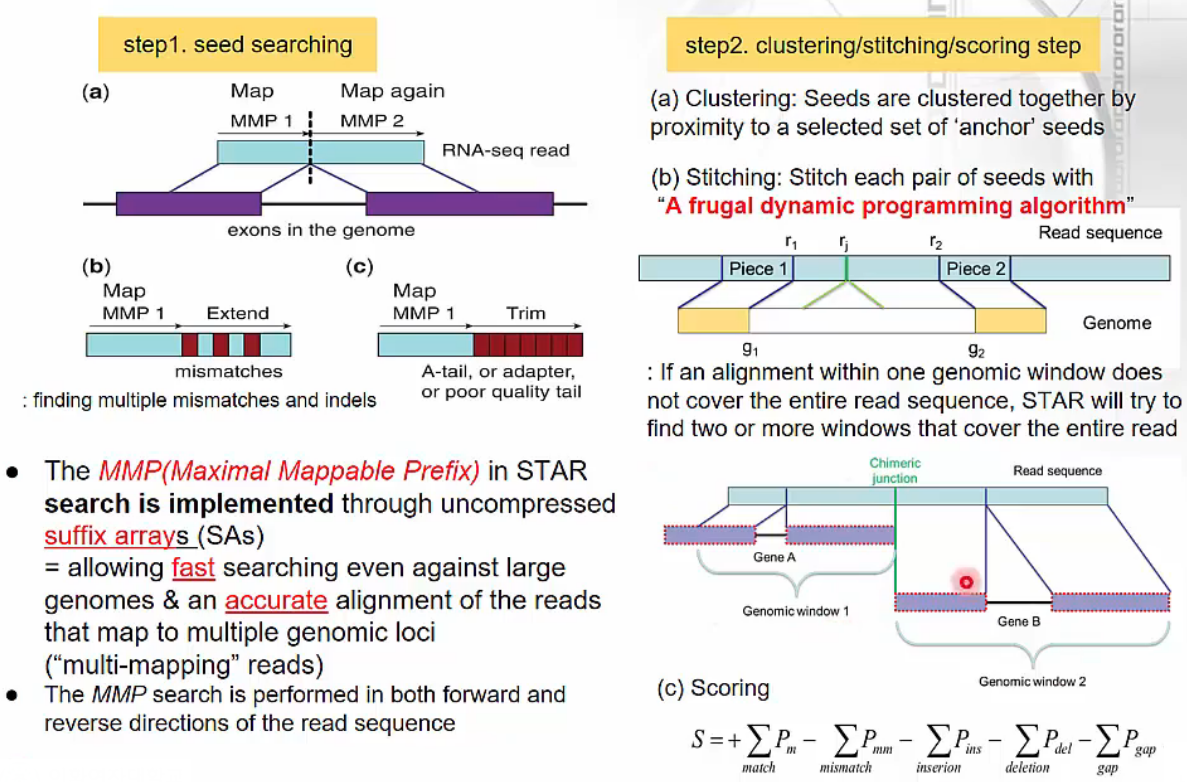 STAR: spliced transcripts alignment to a reference
STAR: spliced transcripts alignment to a reference
https://www.edwith.org/transcriptome/lecture/1382678
- STAR - spliced transcripts alignment to a reference
- step1. seed searching
- Suffiix array 알고리즘을 사용하여 MMP(Maximal Mappable Prefix)를 찾습니다.
- N-mer의 seed reads가 mapping되면 extend하면서 read를 연장합니다.
- setp2. clustering/stiching/scoring step
- clustering: seed reads를 모아 cluster를 만듭니다.
- stitching: frugal dynamic programming altorithm을 사용하여 각 pair of seeds를 연결합니다. 이 때 두 개 이상의 window를 사용하는데 scoring하여 가장 적합한 position을 찾습니다.
- step1. seed searching
- TopHat vs STAR
- TCGA RNA-Seq AML data를 사용하여 비교한 결과입니다.
- 동일한 data의 mapping에 TopHat2는 480분, STAR는 27분 소요됐습니다. STAR가 월등히 빠른 것을 알 수 있습니다.
- 하지만 정확도는 TopHat2가 약간 더 앞선 결과를 보였습니다.
- STAR를 사용하는 것이 더 효율적인 방법임을 생각해 볼 수 있습니다.
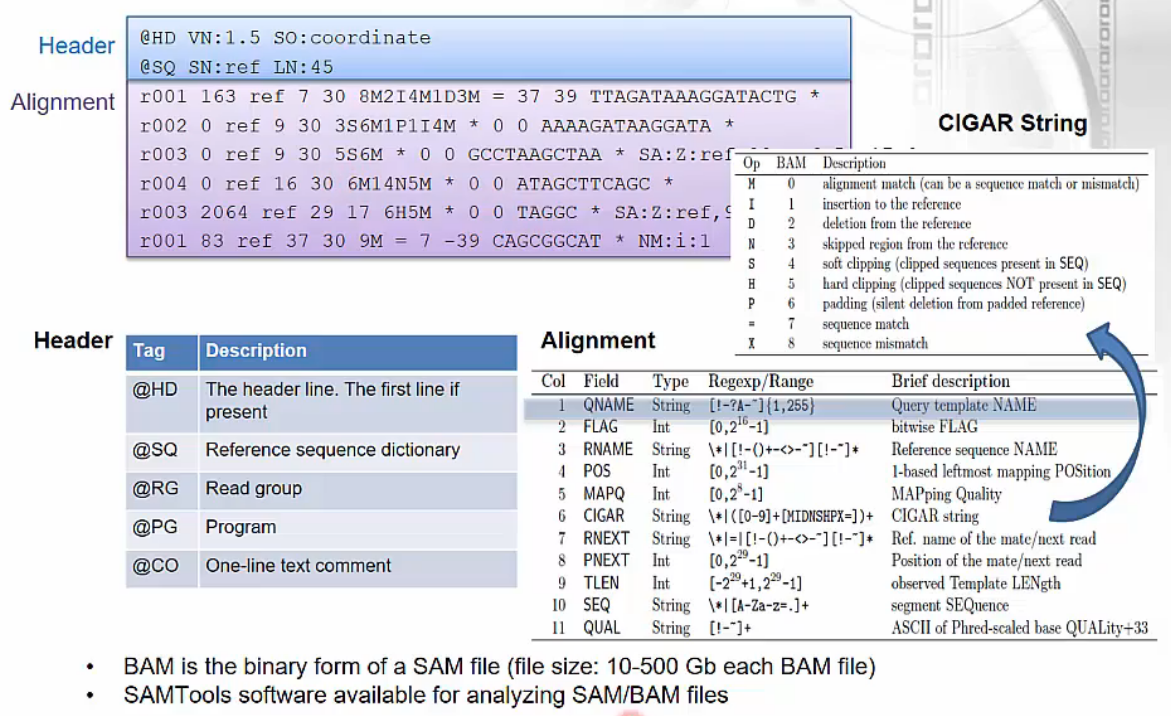 SAM/BAM
SAM/BAM
https://www.edwith.org/transcriptome/lecture/1382678
- Alignment Data Format (SAM/BAM)
- mapping 결과로 나오는 output 파일입니다.
- 6th column의 CIGAR string은 mapping 결과를 요약해서 보여주는데, RNA-Seq data에서 N은 intron 영역을 의미합니다.
- 예) 5M14N8M: exon 5bp + intron 14bp + exon 8bp
Normalization
- Normalization이 필요한 이유는, raw data가 mRNA의 concentration이 아닐 수 있기 때문입니다. 다양한 이유가 존재합니다.
- Sample preparation과 관련된 이슈가 있습니다.
- tissue contamination
- RNA degradation
- amplification efficiency
- reverse transcription efficiency
- microarrays와 관련된 이슈가 잇습니다.
- hybridization efficiency and specificity
- image segmentation
- signal quantificaion
- ‘background’ correction
- RNA-Seq과 관련된 이슈가 있습니다.
- uneven depth of coverage
- uncertainties in mapping and quantification
- Sample preparation과 관련된 이슈가 있습니다.
 Normalization
Normalization
https://www.edwith.org/transcriptome/lecture/1382678
- Normalization은 데이터의 분포와 scale을 조정하여 데이터간 비교 가능하도록 만드는 과정입니다.
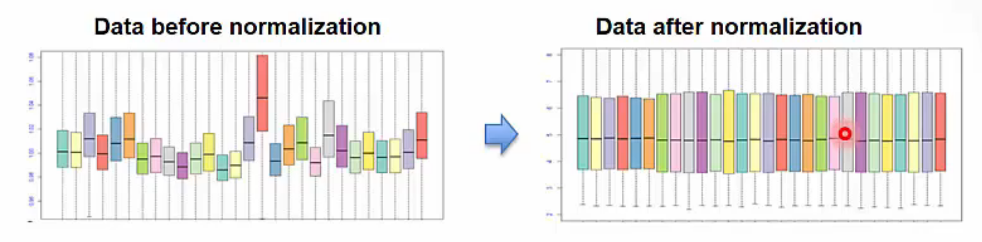
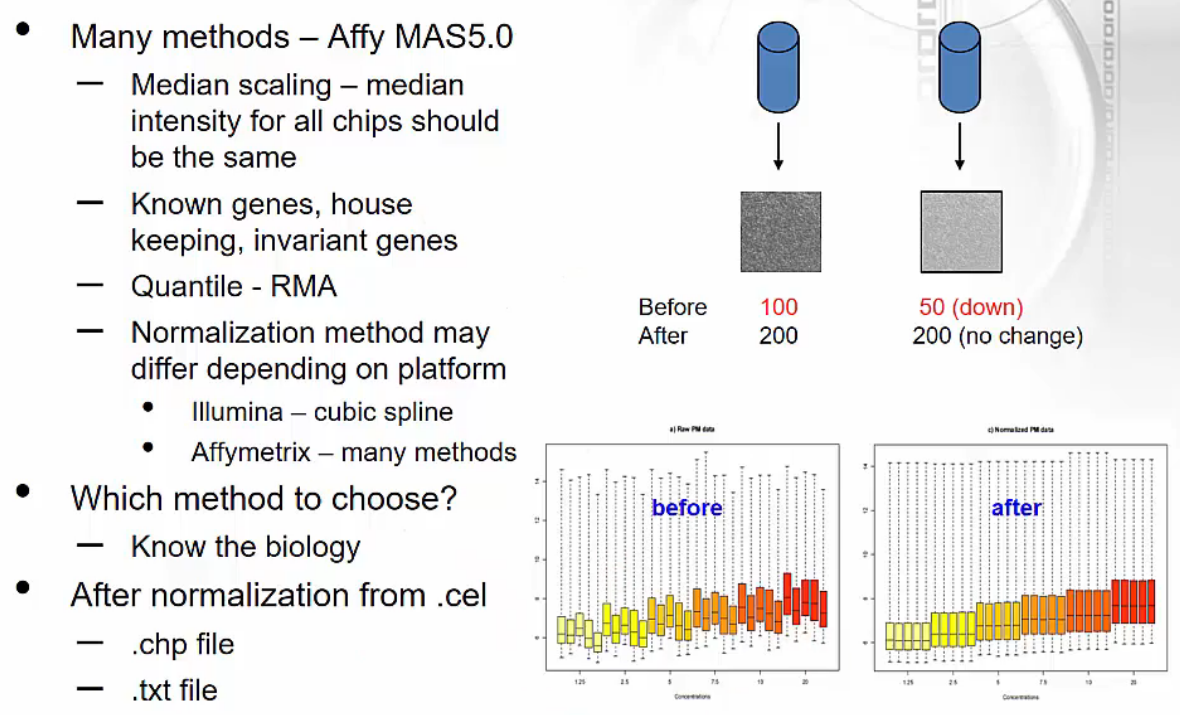 Normalization for microarray data
Normalization for microarray data
https://www.edwith.org/transcriptome/lecture/1382678
- Microarray data의 normalization은 여러 가지 방법이 존재합니다.
- 오른쪽 아래 그림을 보면 실제 duplicate였던 색상별 reads가 normalization 이후 동일한 수준으로 변경되었음을 확인할 수 있습니다.
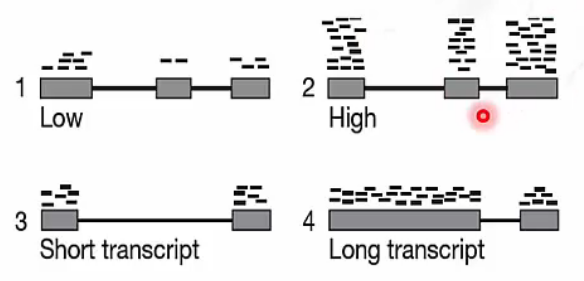 Normalization for RNA-Seq data
Normalization for RNA-Seq data
https://www.edwith.org/transcriptome/lecture/1382678
- RNA-Seq data를 normalization 할 때 두 가지 사항을 고려해야 합니다.
- 1-2: Sequencing depth가 높을수록(2) mapped reads도 증가합니다.
- 3-4: transcript length가 길수록(4) mapped reads도 증가합니다.
따라서 sequencing depth, transcript length에 대해 normalization이 필요합니다.
- RPKM
- Reads Per Kilobase of transcript per Million mapped reads
- reads 백만개 당 kilobase transcript에 mapping된 reads 수를 의미합니다.
- normalization order: depth first -> then length
- FPKM
- Fragments Per Kilobase …
- Paired-end sequencing을 했을 때 하나의 fragment에 대해 forward, reverse 두 번을 sequencing 하는데, 이를 한 개로 간주하고 계산한 결과입니다.
- TPM
- Transcripts Per Million
- normalization order: length first -> then depth
- TPM values can be compared between different samples directly because the sum of all TPMs in each sample art the same
- 지금은 TPM이 정석처럼 사용되고 있습니다.
Quanltification
 Read Counting Rules
Read Counting Rules
https://www.edwith.org/transcriptome/lecture/1382678
- gene 하나의 uniquely mapping되는 read가 있지만 복수 개의 genes에 걸쳐서 mapping되는 경우도 있습니다.
- 다음과 같은 방법으로 이러한 문제를 처리합니다.
- Estimating using unique reads (old)
- Uniquely mapped reads만 사용하는 방식입니다.
- 상대적으로 많은 reads를 버려야하는 단점이 잇습니다.
- tool: NEUMA
- Maximum Likelihood Estimation (MLE)
- low level expression은 정확하게 측정되지 않는 단점이 있습니다.
- tool: EMSAR, MISO, Cufflinks
- Expectation-Maximization
- 현재 가장 많이 사용하는 방식입니다.
- tool: RSEM, eXpress
- Estimating using unique reads (old)
RNA-Seq Analysis Pipeline
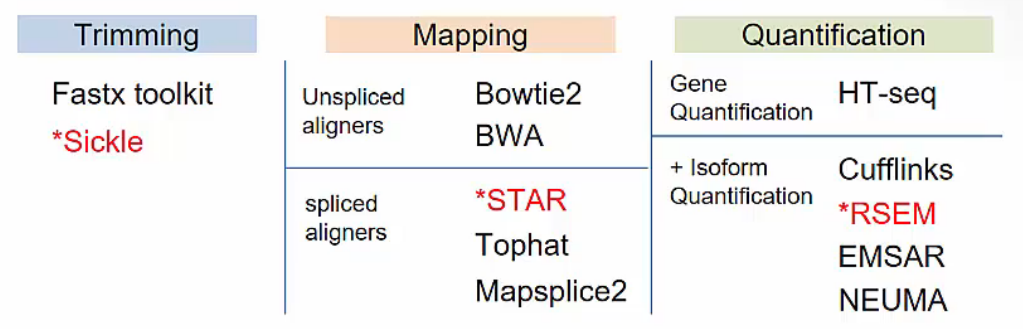 STAR-RSEM Anlysis Pipeline
STAR-RSEM Anlysis Pipeline
https://www.edwith.org/transcriptome/lecture/1382678
- Sickle / STAR / RSEM 을 사용한 RNA-Seq analysis pipeline 입니다.
- Trimming: Sickle
- Mapping STAR v2.6.0c
- Quantification: RSEM v1.3.0
- Differential expression analysis: R v3.6.0 (package: edgeR, preprocessCore, gplots, RColorbrewer)
- Fusion analysis: STARfusion v1.6.0
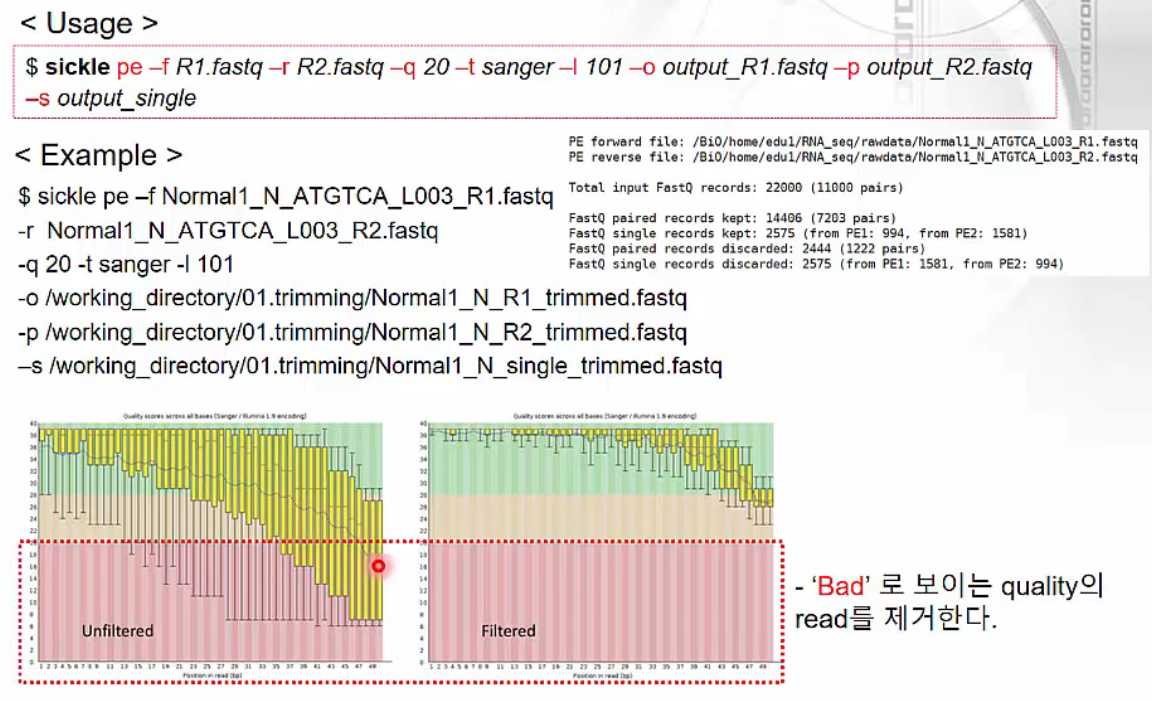 STAR-RSEM Anlysis Pipeline: trimming
STAR-RSEM Anlysis Pipeline: trimming
https://www.edwith.org/transcriptome/lecture/1382678
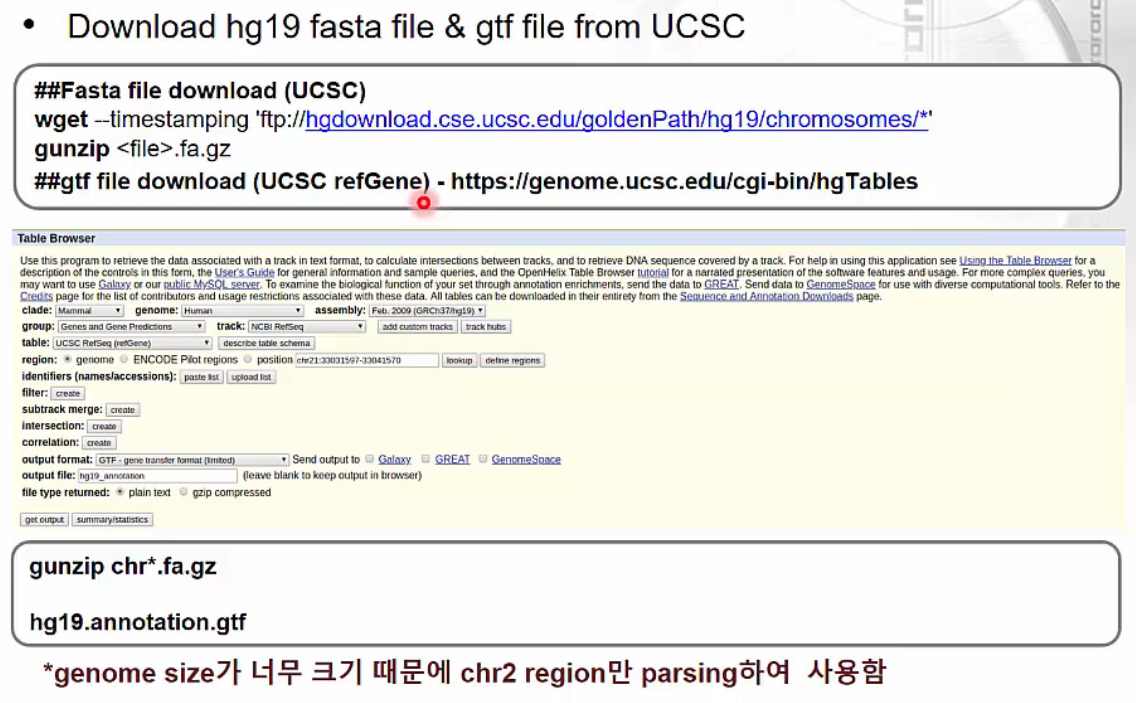 STAR-RSEM Anlysis Pipeline: preparing reference
STAR-RSEM Anlysis Pipeline: preparing reference
https://www.edwith.org/transcriptome/lecture/1382678
 STAR-RSEM Anlysis Pipeline: mapping & quantification
STAR-RSEM Anlysis Pipeline: mapping & quantification
https://www.edwith.org/transcriptome/lecture/1382678
 STAR-RSEM Anlysis Pipeline: mapping & quantification
STAR-RSEM Anlysis Pipeline: mapping & quantification
https://www.edwith.org/transcriptome/lecture/1382678
 STAR-RSEM Anlysis Pipeline: mapping & quantification
STAR-RSEM Anlysis Pipeline: mapping & quantification
https://www.edwith.org/transcriptome/lecture/1382678
Take Home Message
RNA-Seq data preprocessing 과정에 대해 배울 수 있었습니다. STAR-RSEM pipeline이 가장 많이 사용되고 있음을 알 수 있었습니다.