Variants Types
DNA 서열에서 다양한 종류의 변이가 나타날 수 있으며, 일부 변이는 암, 유전성 질환과 연관되어 있습니다.
| Variants Types | Description |
|---|---|
| Substitution - Missense | Coding region에서 nucleotide의 substitution으로 나타나는 variants. 그 결과 region이 coding하는 amino acid가 다른 amino acid로 바뀜 |
| Substitution - Nonsense | Coding region에서 nucleotide의 substitution으로 나타나는 variants. 그 결과 region이 coding하는 amino acid가 stop codon으로 바뀌고 protein의 기능에 영향을 줌 |
| Insertion | 한 개 이상의 nucleotides가 추가된 variants. 그 결과 protein이 적절한 기능을 하지 못하도록 영향을 줄 수 있음 |
| Deletion | 적어도 한 개 이상의 nucleotides가 삭제된 variants. 그 결과 protein이 적절한 기능을 하지 못하도록 영향을 줄 수 있음 |
| Deletion-Insertion(delins), Insertion-Deletion(indel) | Deletion과 Insertion이 같은 시간 같은 region에서 일어난 variants. 그 결과 protein이 적절한 기능을 하지 못하도록 영향을 줄 수 있음 |
| Duplication | 한 개 이상의 nucleotides가 복제되거나 반복되어 나타나는 variants. 그 결과 protein이 적절한 기능을 하지 못하도록 영향을 줄 수 있음 |
| Inversion | Original sequence 상에서 한 개 이상의 nucelotides가 reverse 방향으로 대체된 variants. |
| Frameshift | 한 개 이상의 nucleotides가 추가되거나 삭제되어 codon 서열을 변경시키는 variants. 그 결과 protein이 적절한 기능을 하지 못하도록 영향을 줄 수 있음 |
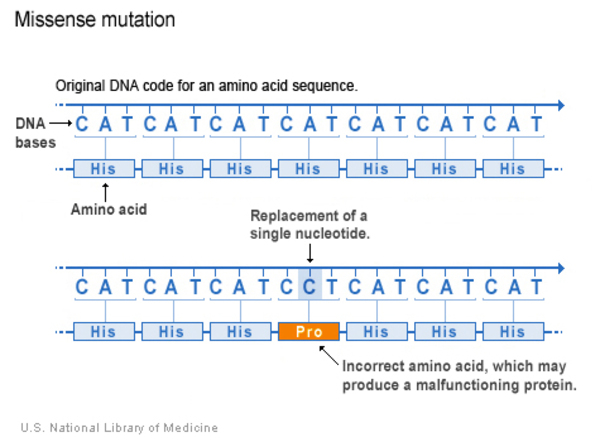 Substitution - Missense
Substitution - Missense
https://medlineplus.gov/genetics/understanding/mutationsanddisorders/possiblemutations/
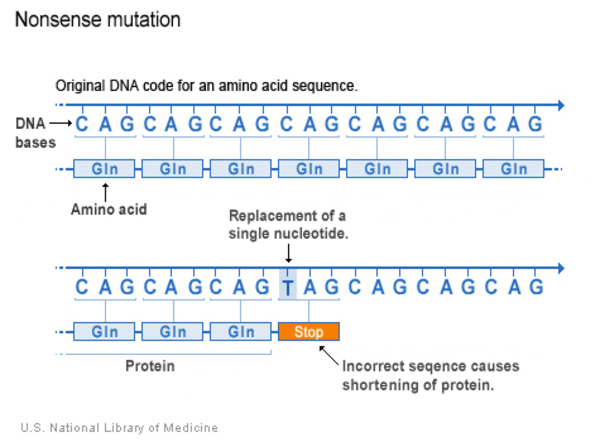 Substitution - Nonsense
Substitution - Nonsense
https://medlineplus.gov/genetics/understanding/mutationsanddisorders/possiblemutations/
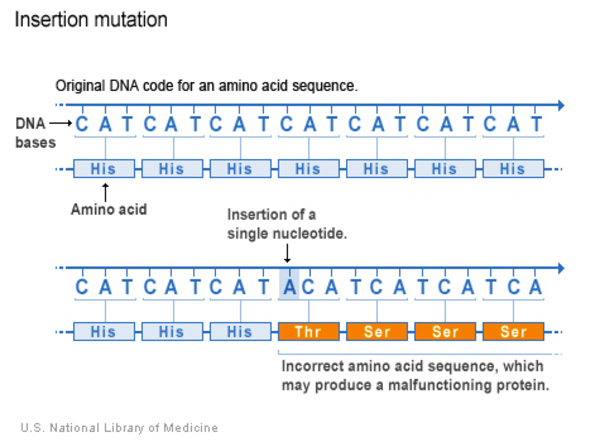 Insertion
Insertion
https://medlineplus.gov/genetics/understanding/mutationsanddisorders/possiblemutations/
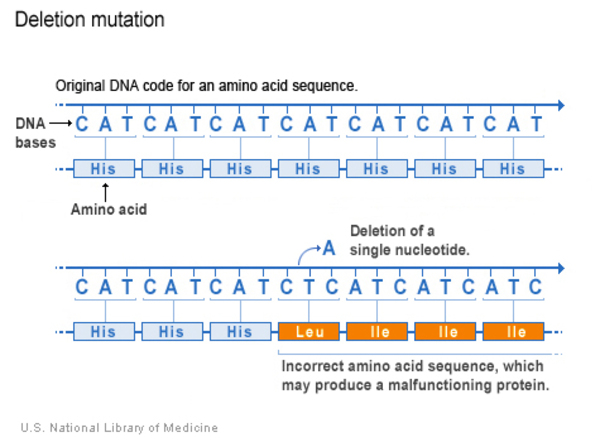 Deletion
Deletion
https://medlineplus.gov/genetics/understanding/mutationsanddisorders/possiblemutations/
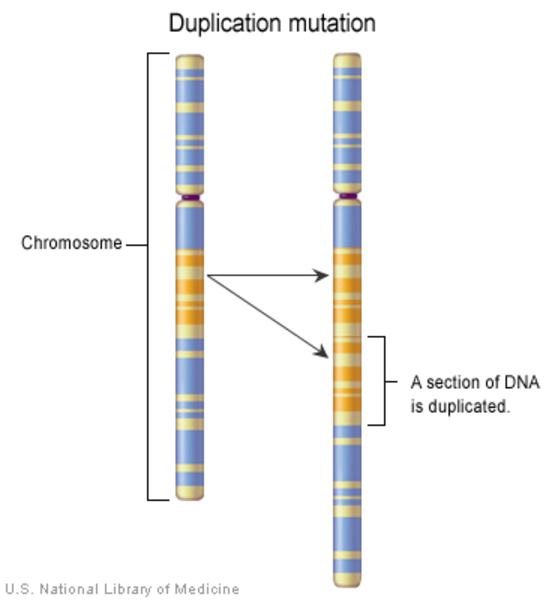 Duplication
Duplication
https://medlineplus.gov/genetics/understanding/mutationsanddisorders/possiblemutations/
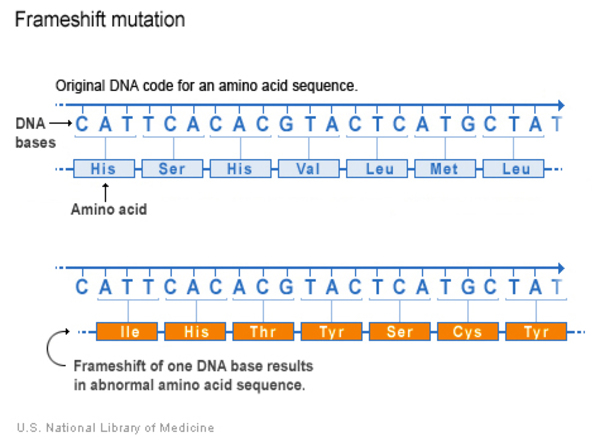 Frameshift
Frameshift
https://medlineplus.gov/genetics/understanding/mutationsanddisorders/possiblemutations/
Variants Error
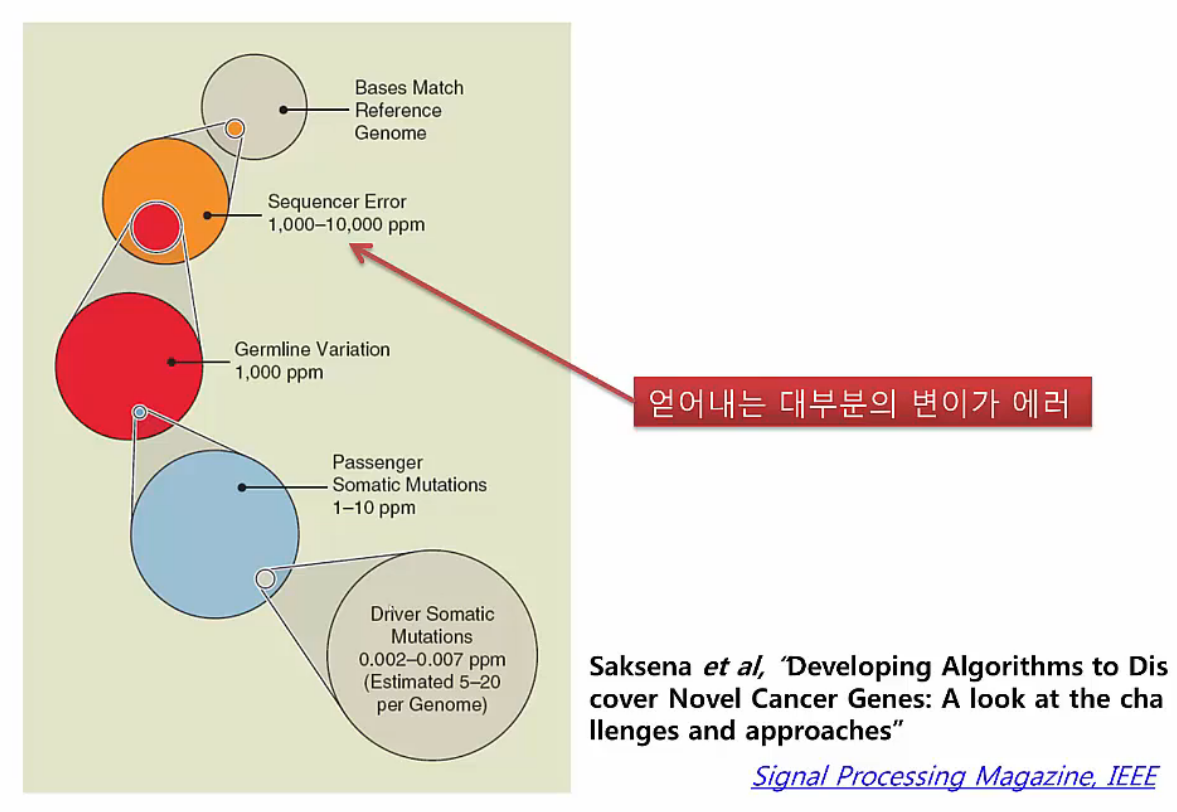 Variants Error
Variants Error
https://www.edwith.org/ngs-data-variation/lecture/1382349?isDesc=false
Reference genome과 다른 bases로 확인된 variants의 대부분이 사실 sequencer error입니다. 여기에서 최종 driver somatic mutations로 남는 variants를 선별하는 것은 까다로운 과정입니다. 따라서 처음에 called variants에서 sequencer error를 찾고 제거하는 과정은 매우 중요합니다.
Sequencer error를 제거하기 위해서 특정 region을 여러 번 반복해서 sequencing 합니다. 이를 정량화한 값을 read depth라고 부르는데, 특정 영역을 지나는 read의 수를 count한 값 입니다. 이처럼 특정 region을 반복해서 시퀀싱하면 sequencer error가 발견되더라도 이를 error로 분류할 가능성이 높아집니다.
또한 basecall error를 정량화한 수치인 phred score를 사용하여 sequencer error를 찾아낼 수 있습니다. 이 수치는 basecall이 틀렸을 확률 e에 대해 $-10log_{10}$을 대입한 값을 의미합니다. 10, 20, 30, 40 값이 각각 basecall 틀렸을 확률 10%, 1%, 0.1% 0.01%를 의미합니다.
Mapping error를 정량화한 수치인 MapQ를 사용하여 mapping error를 가늠해 볼 수 있습니다. 마찬가지로 mapping이 틀렸을 확률 e에 대해 $-10log_{10}$을 대입한 값을 의미합니다. 예를 들어 하나의 read가 여러 곳에 mapping 될 수 있다면 e가 증가하는 등 여러가지 근거에 기반하여 MapQ를 계산합니다.
Variants Calling
Variants error를 확인할 수 있는 수치들을 기반으로 sequencing 결과 관찰된 variants가 true positive인지 error인지 판단합니다. 아래와 같이 reference sequence가 T인 position에서 다섯 개의 read가 mapping된 상황을 가정해 봅니다. 네 개 read는 T, 한 개 read는 G이고 각각 basecall quality와 mapping quality가 표기되어 있습니다.
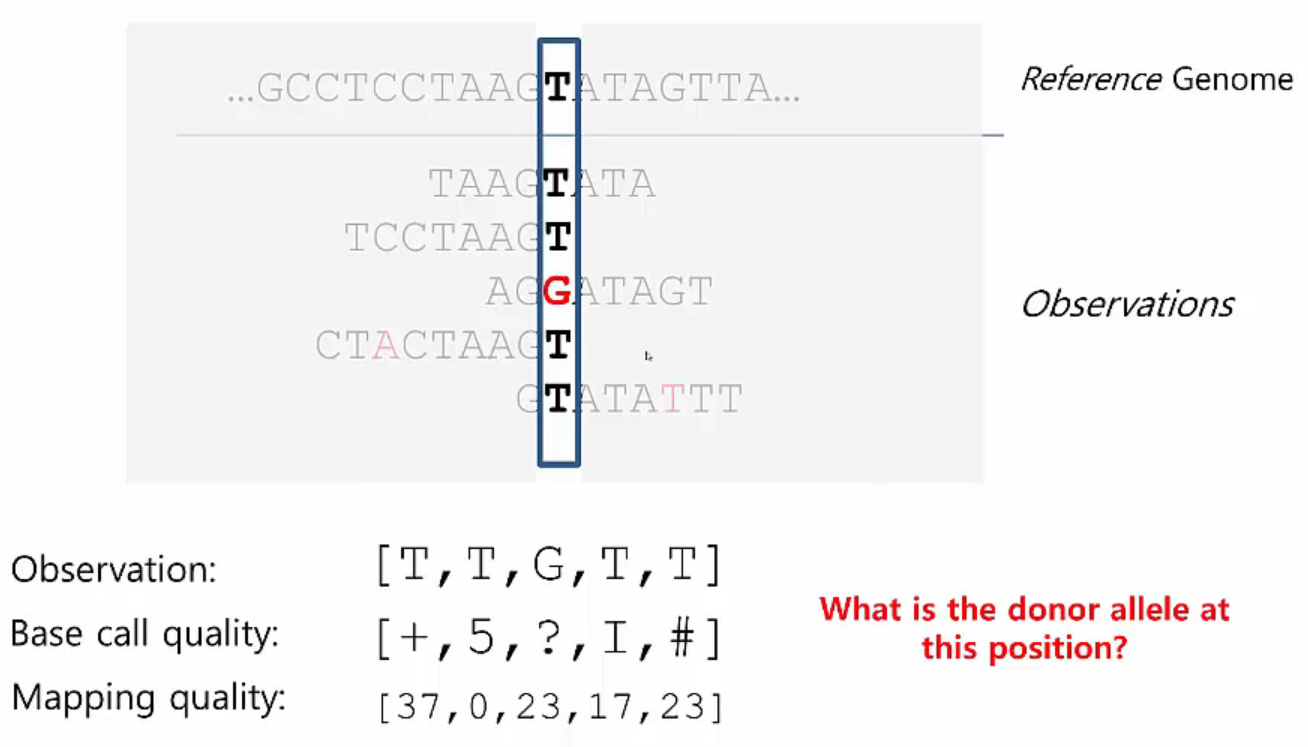 Variants Calling
Variants Calling
https://www.edwith.org/ngs-data-variation/lecture/1382349?isDesc=false
사람의 genome은 diplod이므로 하나의 position에서 두 개의 allele이 존재합니다. A, T, C, G 네 개의 base가 있으므로 경우의 수를 따지면 총 16가지 조합이 가능합니다. Reference sequence와 동일할 때 A, 동일하지 않을 때 B라고 지칭하면 경우의 수는 4가지 조합으로 단순화 할 수 있습니다.
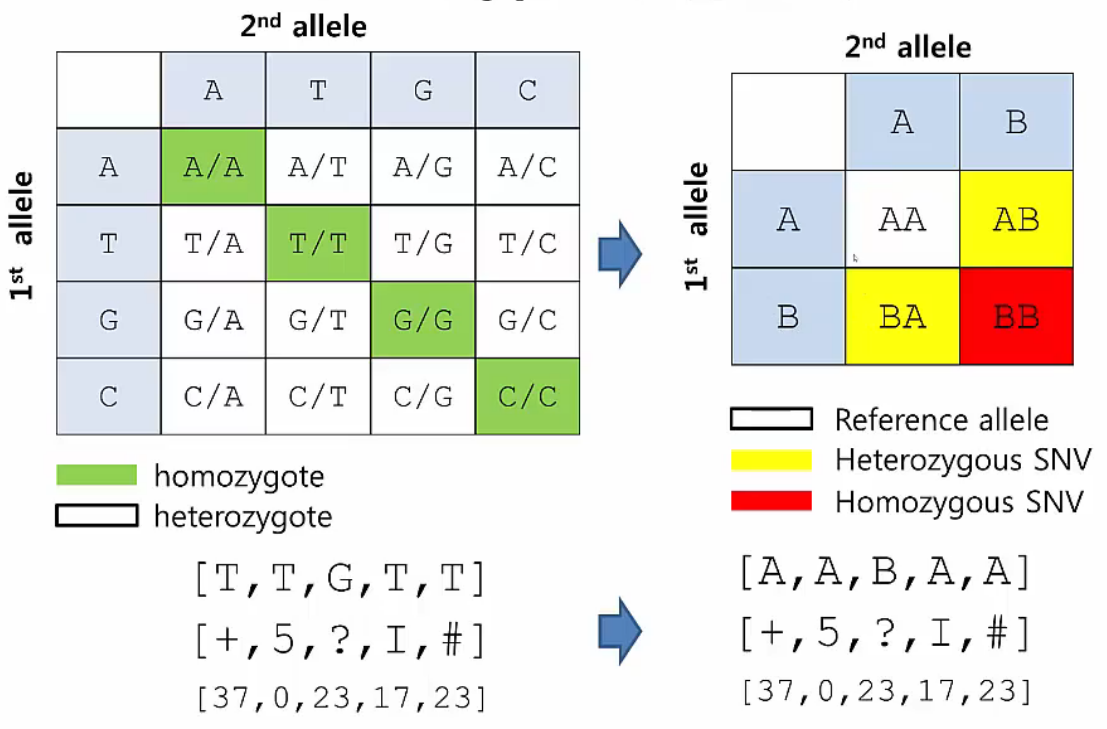 Simplify Genotyping
Simplify Genotyping
https://www.edwith.org/ngs-data-variation/lecture/1382349?isDesc=false
다시 처음에 가정한 상황으로 돌아와서 genotyping을 단순화 했습니다. 이제 B가 true positive인지, 혹은 error로 인한 false positive인지 검증하는 단계입니다.
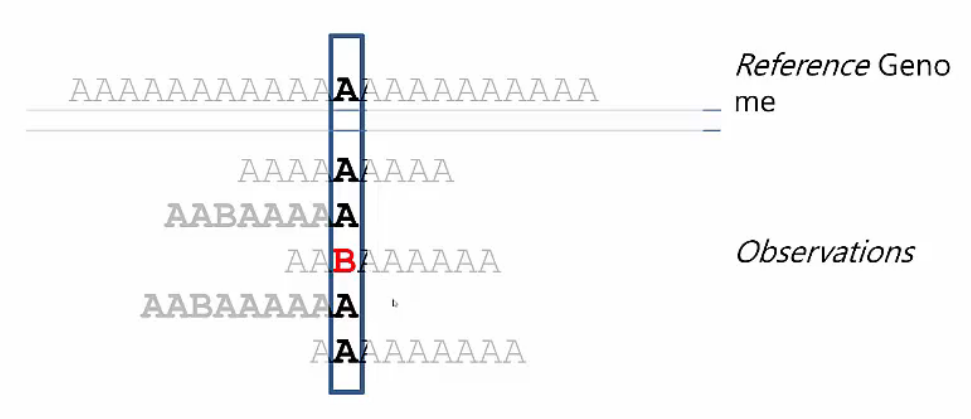 Simplify Genotyping
Simplify Genotyping
https://www.edwith.org/ngs-data-variation/lecture/1382349?isDesc=false
여기부터 확률 개념이 사용됩니다. 사람의 유전체는 diploid이므로 해당 위치의 genotype은 AA or AB or BB 중 하나가 될 수 있습니다. Pr(G=AA) or Pr(G=AB) or Pr(G=BB) 중 가장 확률이 높은 genotype으로 추론하는 것이 바람직 합니다. 이 과정에 sqeuncing data를 활용할 수 있습니다. 즉, data가 주어졌을 때 각 genotype을 얻을 수 있는 확률은 Pr(G=AA|D) or Pr(G=AB|D) or Pr(G=BB|D)로 계산할 수 있습니다. 확률을 바로 계산하기 어려우므로 bayes rule을 활용합니다.
| 위 식에서 $$ Pr(D | G) $$를 계산해 봅시다. 즉, genotype이 주어진 상태에서 data가 확인될 확률을 구하는 것입니다. |
첫 번째 자리의 base는 ‘A’이며 base quality는 ‘+’입니다.
Genotype을 ‘AA’라고 가정할 때, ‘A’가 sequencing될 확률은, P(1-e) 입니다.
Genotype을 ‘AB’라고 가정할 때, ‘A’가 sequencing될 확률은 error없이 ‘A’가 읽힐 확률 \(\frac{1}{2}*P(1-e)\)와 ‘B’가 error에 의해 ‘A’로 읽힐 확률 \(\frac{1}{2}*\frac{1}{4}*P(e)\)를 더한 값 입니다.
Genotype을 ‘BB’라고 가정할 때, ‘A’가 sequencing될 확률은 error에 의해 ‘A’로 읽힐 확률 \(\frac{1}{4}*P(e)\)입니다.
세 번째 자리의 base ‘B’에 대한 Pr(D|G)도 마찬가지로 계산할 수 있습니다.
 Calculation of Pr(D|G)
Calculation of Pr(D|G)
https://www.edwith.org/ngs-data-variation/lecture/1382349
Take Home Message
NGS DNA variants 종류와 genotyping logic을 이해할 수 있었습니다.