본 post는 국가생명연구자원정보센터(KOBIC) 주관 연세대학교 의과대학 김상우 교수님의 NGS 데이터 분석 기초 강의를 정리한 내용입니다.
Intro
NGS를 이용한 somatic mutation 탐지 방법을 알아봅니다. 또한 low allele frequency mutation, mosaic mutation과 같이 검출하기 어려운 mutation에 대해 알아봅니다.
Germline vs Somatic Mutation
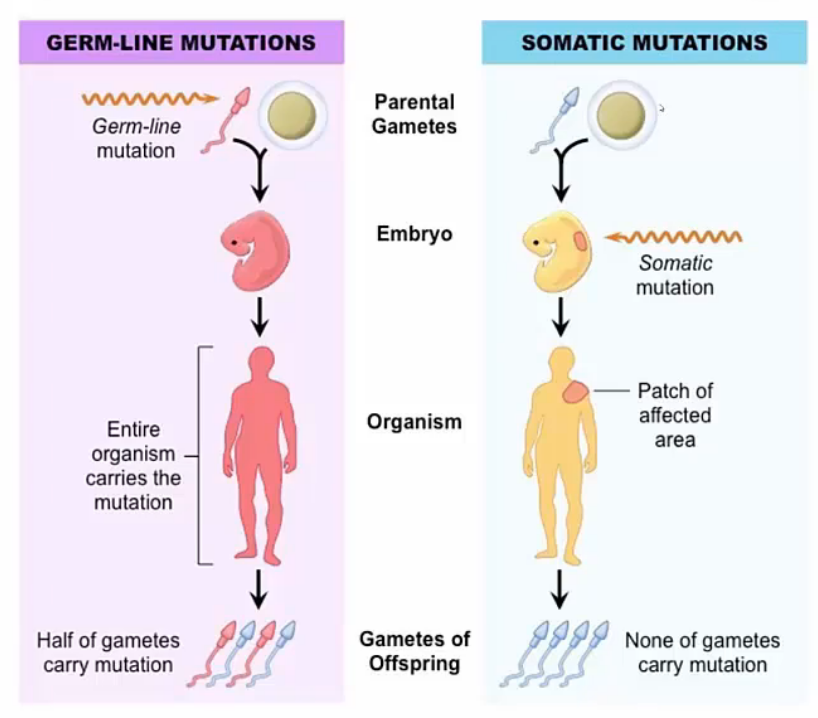 Germline vs Somatic Mutation
Germline vs Somatic Mutation
Germline mutation은 생식세포에서 발생한 mutation으로 사람이 성장하면서 모든 cell이 mutation을 공유합니다.
반면 Somatic mutation은 체세포에서 발생한 mutation으로 사람의 일부 cell만 mutation을 가지고 있습니다.
Variant Allele Frequency
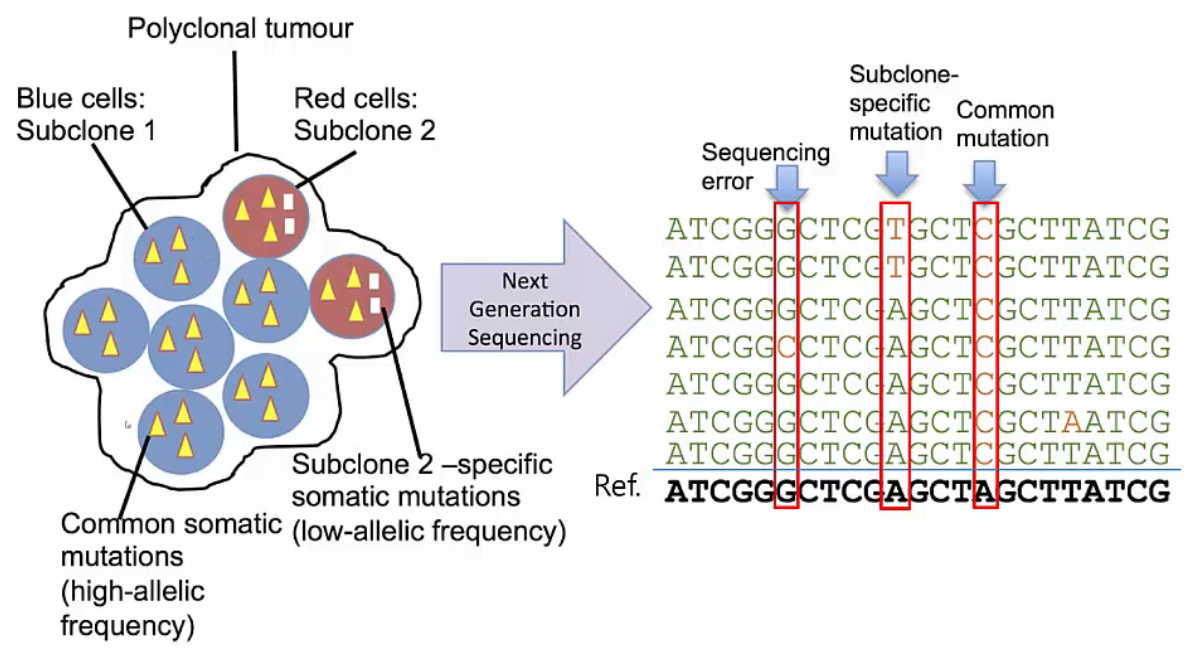 Variant Allele Frequency
Variant Allele Frequency
Tumour가 발달하면서 여러 개의 subclone으로 나뉠 수 있습니다. 그림에서 subclone1과 subclone2에서 공통적으로 나타나는 노란색 mutation은 common somatic mutation으로 high-allelic frequency를 보입니다. 반면에 subclone2에서만 나타나는 하얀색 mutation은 specific somatic mutation으로 low-allelic frequency를 보입니다.
해당 tumour를 sequencing 했을 때 read의 구성은 오른쪽 그림과 같습니다. Common mutation은 모든 read에서 확인되는 반면, subclone-specific mutation은 일부 read에서만 확인됩니다.
Somatic Mutation Analysis: JointSNVMix
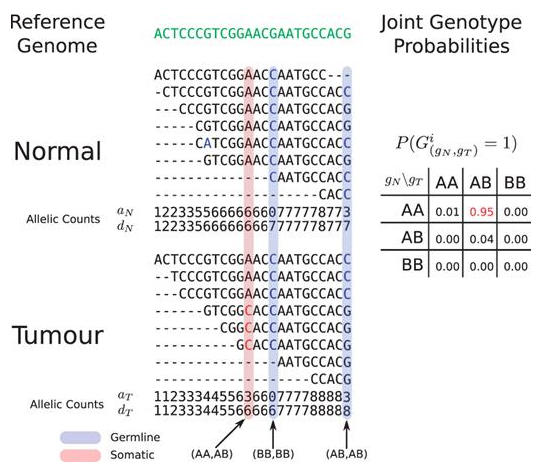 JointSNVMix
JointSNVMix
https://doi.org/10.1093/bioinformatics/bts053
University of British Columbia의 Shah Lab에서 제안한 Joint Genotype Prbability로 somatic mutation analysis 방법 중 하나입니다. (Andrew Roth et al., JointSNVMix : A Probabilistic Model For Accurate Detection Of Somatic Mutations In Normal/Tumour Paired Next Generation Sequencing Data)
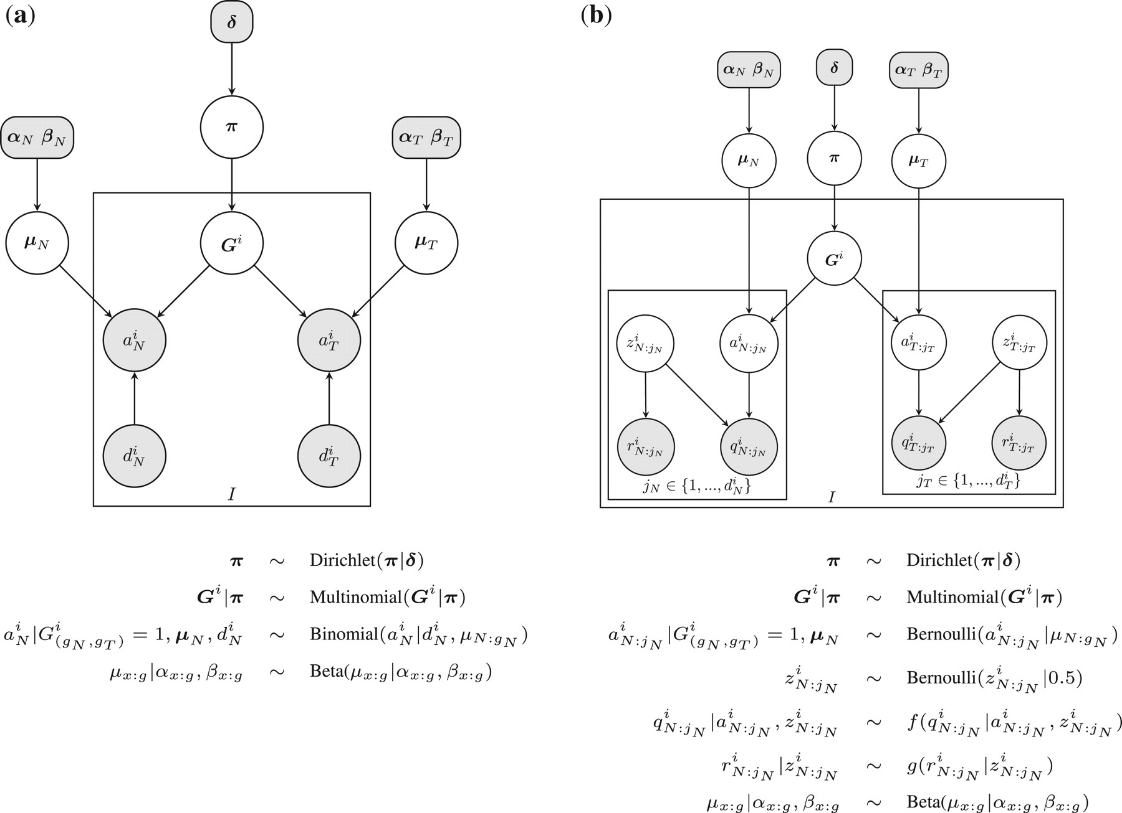
동일한 position에서 tumour와 matched normal의 allele을 모두 고려하여 somatic mutation을 찾습니다. 사람은 diploid이며 이론상 하나의 position에서 나올 수 있는 genotype은 A, B만 있다고 가정하면, 가능한 genotype은 AA, AB, BB입니다. Tumour와 normal에서 총 아홉 가지의 genotype 조합이 가능합니다. 그리고 각 조합이 의미하는 바는 아래와 같습니다. Somatic mutation은 (AA,AB), (AA,BB)인 경우입니다.
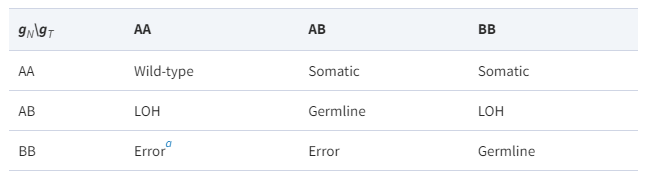 JointSNVMix
JointSNVMix
https://doi.org/10.1093/bioinformatics/bts053
각 position에서 allele을 count한 다음 아래와 같이 두 가지 확률 모델을 사용하여 확률적으로 가능성이 높은 genotype을 결정합니다. 각 node에 대한 설명은 논문(Andrew Roth et al., JointSNVMix : A Probabilistic Model For Accurate Detection Of Somatic Mutations In Normal/Tumour Paired Next Generation Sequencing Data)을 참고해 주세요.
 JointSNVMix
JointSNVMix
https://doi.org/10.1093/bioinformatics/bts053
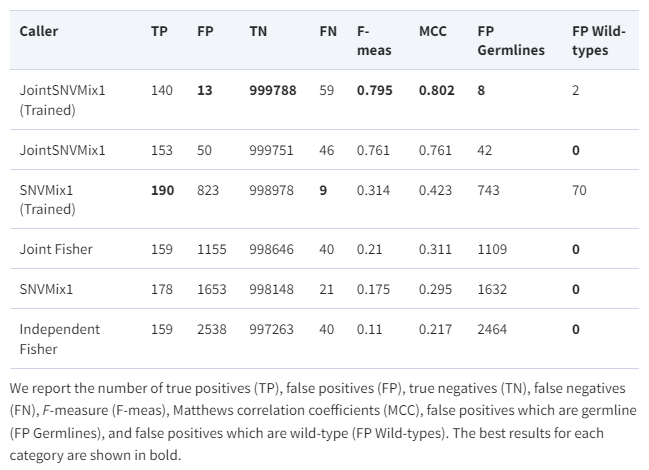
마지막으로 JointSNVMix의 성능평가 결과입니다. 그 외 caller들은 성능평가를 위해 변형하거나 사용한 다른 방식의 somatic mutation analysis입니다. JointSNVMix가 F-measure, MCC(Matthews Correlation Coefficient)에서 가장 우수한 결과를 보입니다.
 JointSNVMix
JointSNVMix
https://doi.org/10.1093/bioinformatics/bts053
Somatic Mutation Analysis: MuTect
 MuTect2
MuTect2
https://doi.org/10.1093/bioinformatics/bts053
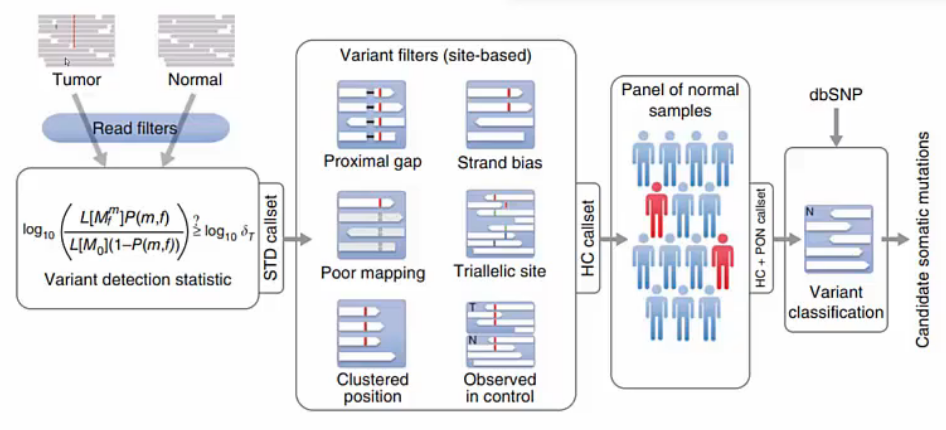
MuTect2는 Broad Institute에서 개발한 somatic variants caller입니다. Baysian classifier를 사용하며 대상 position에서 variant가 없다고 가정($M_{0}$)하고 구한 확률과 varinat가 있다고 가정($M_{f}^{m}$)하고 구한 확률을 비교하여 더 가능성이 높은 쪽을 택하는 방식입니다. 이후에 다양한 방식의 variant filters를 거쳐 FP를 제거하고, normal 사람들을 sequencing하여 얻은 변이(MAF)를 제거한 뒤 candidate somatic mutations를 추립니다. br>
저빈도 변이와 모자이크 변이
 Mosaic Variants
Mosaic Variants
https://doi.org/10.1093/bioinformatics/bts053
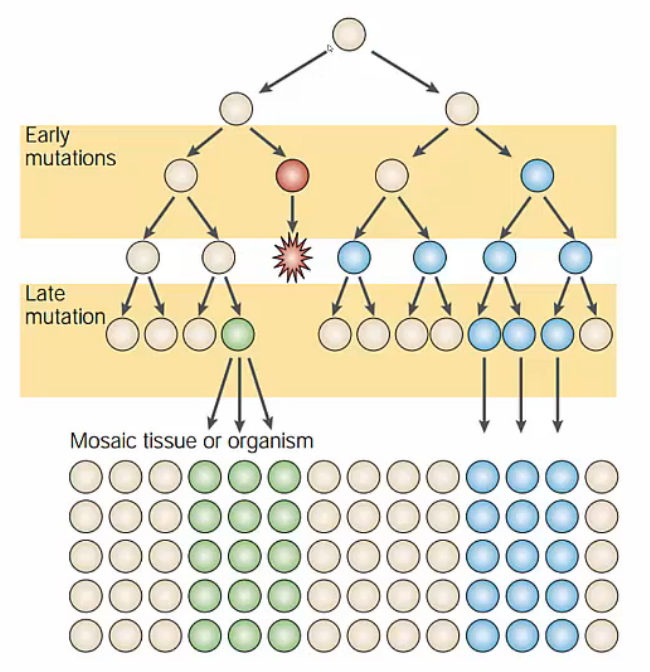
Mosaic은 한 개체 또는 한 지역 내에 서로 다른 유전형이 함께 공존하는 것을 의미합니다. 비교적 이른 시기에 발생한 mutation은 많은 세포가 mutation을 공유하게되고, 비교적 늦은 시기에 발생한 mutation은 상대적으로 일부 세포만 mutation을 공유하게 됩니다.
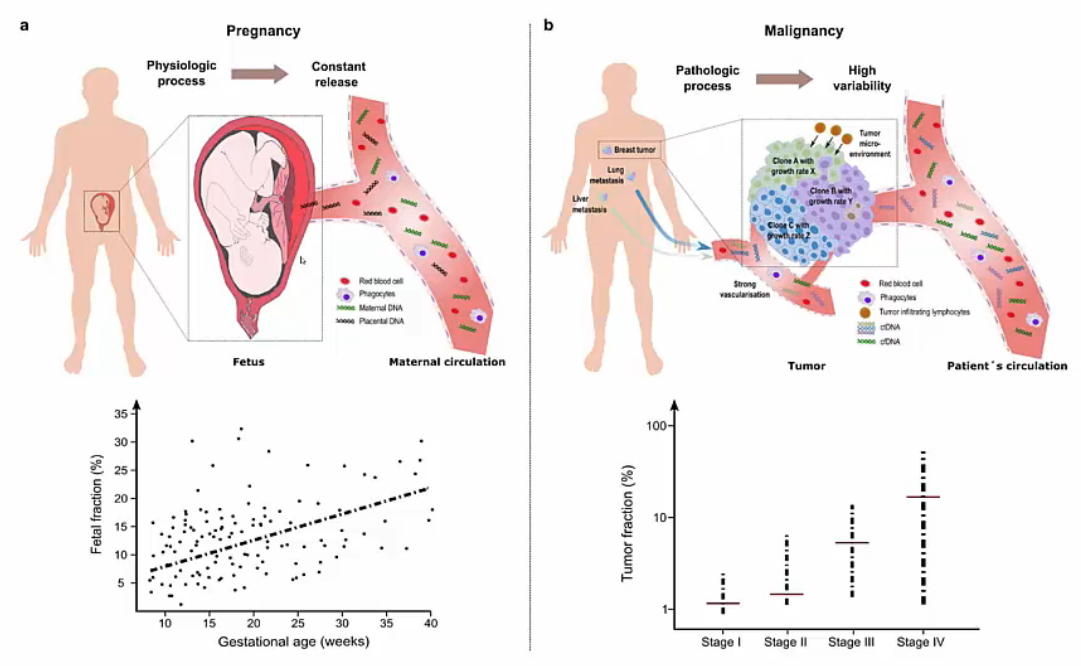 Liquid Biopsy
Liquid Biopsy
https://doi.org/10.1093/bioinformatics/bts053
혈액 내 미세하게 떠다니는 DNA를 뽑아서 sequencing하고 mutations를 확인하는 기술입니다. 일례로 산모의 혈액에는 태아의 DNA가 떠다니는데 이를 뽑아서 genotype을 확인하여 태아의 건강을 확인합니다.
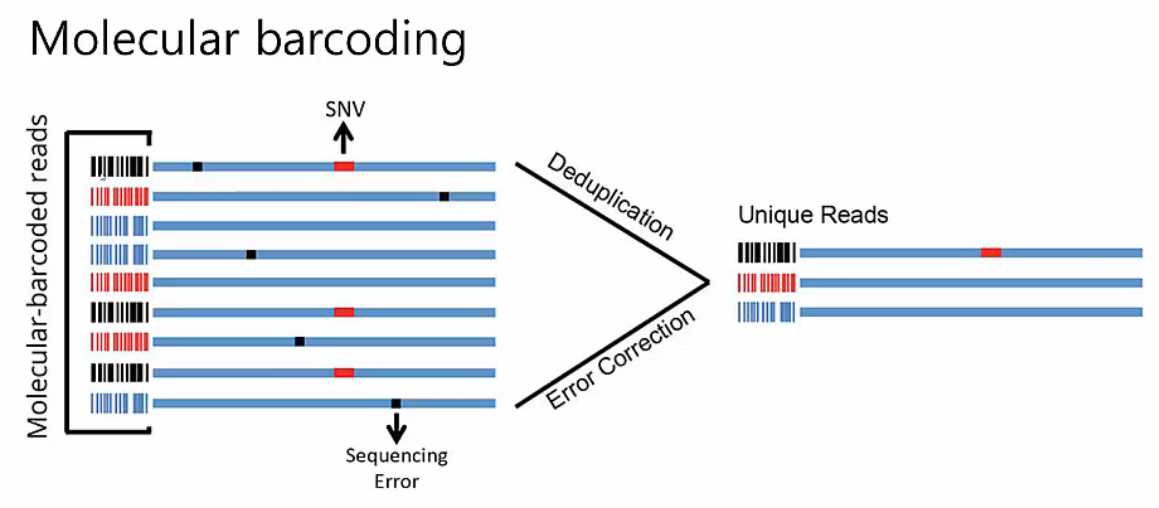 Unique Molecular Identifier
Unique Molecular Identifier
https://doi.org/10.1093/bioinformatics/bts053
저빈도 변이를 calling할 때 발생하는 error들을 제거하기 위해 UMI를 사용합니다. 원본 DNA fragment마다 고유한 index를 붙이고, duplicate를 제거할 때 동일한 UMI를 가진 duplicate들만 모아서 PCR error로 추정되는 mutation은 제거합니다.
이 밖에도 liquid biopsy는 variant 찾는 것이 매우 어려우므로 다양한 기술이 개발 중에 있습니다.
Take Home Message
Somatic variants call 과정과 저빈도, 모자이크 변이에 대해 이해할 수 있었습니다.